 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ANEEQA NOOR | + 1134 word(s) | 1134 | 2021-03-16 08:02:35 | | | |
| 2 | Nicole Yin | Meta information modification | 1134 | 2021-04-06 11:38:41 | | | | |
| 3 | Nicole Yin | Meta information modification | 1134 | 2021-04-06 11:39:12 | | |
Video Upload Options
Neurodegenerative Proteinopathies, also known as protein conformational diseases or amyloidosis, are a group of diseases associated with the deposition of misaggregated proteins in the nervous system.
1. Introduction
Most of the common neurodegenerative disorders—Alzheimer’s disease (AD), Parkinson’s disease (PD), Creutzfeldt–Jacob disease (CJD), Dementia with Lewy bodies (DLB), Huntington disease (HD), and Amyloid Lateral Sclerosis (ALS) —are proteinopathies. Together, these diseases affect millions of lives around the world and have devastating economic implications. AD, the most frequently diagnosed among these listed diseases, affects almost one-tenth of the population above 65 years of age[1]. The number of people suffering from these diseases is increasing rapidly with an increase in life expectancy and it is predicted that by 2050, 135.46 million people will be living with various types of neurodegenerative dementias[2]. Despite the attention of the scientific community, these disorders are far from resolved. The patients can be treated to alleviate the symptoms, but the lack of a cure still means that such a diagnosis can seal their fate.
2. Amyloid
Although proteinopathies present similarities in their pathological mechanisms, the psychological and physiological symptoms of all these disorders vary and depend on the region of the brain affected. A summary of age at onset, primary sites of pathology, and common symptoms of major neurodegenerative proteinopathies is presented in Table 1. These variations, in turn, are dictated by the proteins that are involved in amyloid formation (Table 2).
Table 1. Age at onset, affected brain regions, and common symptoms of major neurodegenerative proteinopathies. Age at onset represents a range, rather than mean, due to multiple clinical variants of each of these disorders. * sporadic CJD.
| Proteinopathy | Age at Onset (Years) | Primary Region | Common Symptoms |
|---|---|---|---|
| AD | 40–65 (early and late-onset variants) |
Hippocampus and entorhinal cortex. | Memory and language impairment and visuospatial deficits. [3][4] |
| PD | 40–50 | Substantia nigra (midbrain). | Rigidity, resting tremor and bradykinesia. [5] |
| sCJD * | 44–70 (depends on subtype) |
Cerebral cortex and cerebellum. | Cognitive impairment and myoclonus. [6] |
| DLB | 50–80 | Midbrain and neocortex. | Parkinsonian syndrome, autonomic and sleep fluctuations and hallucinations. [7] |
| HD | 20–49 | Caudate nucleus and putamen (basal ganglia). | Choreiform movements, emotional and behavioral alterations, bradykinesia. [8] |
| ALS | 45–55 | Motor neurons. | Focal muscle wasting, spasticity and flexor spasms. [8][9] |
Table 2. A summary of the structure and variants of major amyloidogenic proteins associated with neurodegenerative proteinopathies.
| Amyloids | Precursor Protein | Associated Diseases | Proteoforms or Other Variants |
|---|---|---|---|
| Aβ | Amyloid beta A4 protein: Intrinsically disordered protein with 770 residues | AD, Cerebral amyloid angiopathy (CAA)[10][11]. | 26 differentially truncated and post translationally modified proteoforms [12] |
| α-Synuclein | Alpha Synuclein: Intrinsically disordered protein with 140 residues | PD and DLB [13] | 11 differentially truncated and post translationally modified proteoforms [14] |
| PrPSc | Major prion protein: Intrinsically disordered protein with 253 amino acids | CJD, Fatal Familial Insomnia (FFI), Gerstmann-Straussler-Scheinker disease (GSS), Huntington disease-like type 1 (HDL1), Kuru and Spongiform encephalopathy [15] | 2 Proteoforms based on Proteinase-K resistance Genetic variants (codon 129 polymorphism). [16] |
| ASOD | Superoxide dismutase: Intrinsically disordered protein with 154 amino acids | ALS—TDP-43 amyloids also involved. [17][18] | Genetic variants. No proteoforms reported yet. [19] |
| ATau | Microtubule-associated protein tau: Intrinsically disordered protein with 758 amino acids | Frontotemporal dementia (FTD), AD, Progressive Supranuclear Palsy (PSP), Corticobasal degeneration (CBD), Pick’s disease, Argyrophilic grain disease, Dementia with Lewy bodies and Parkinsonism linked to chromosome 17. [20] | Six isoforms. Differentially post translationally modified proteoforms. [21] |
| ATTR | Transthyretin: Mostly β-sheet with 147 amino acids | Familial Amyloid polyneuropathy, Leptomeningeal amyloidosis. [22] | Differentially oxidized proteoforms. [23] |
| AHtt | Huntington: Intrinsically disordered protein with 3142 residues | Huntington disease. [24] | Differentially post translationally modified proteoforms. [25] |
In addition to similarities in the mechanism of propagation, prion-like proteins have also adapted another interesting aspect of PrPSc biology. PrPSc can give rise to several clinical variants of prion diseases. This heterogeneity has been attributed to the existence of distinct PrP strains. Strains are defined as conformers of a specific amyloidogenic protein, in this case PrPSc, that differ with respect to their transmission, brain-lesion profiles, incubation periods, and disease phenotypes along with certain biochemical characteristics like Post-translational modifications, sensitivity to proteinase-K, and electrophoretic mobility. The distinct conformational characteristics of each PrP strain are transmitted into the host where it propagates and causes distinct phenotypes[26]. Codon 129 polymorphism gives rise to at least three known strains of PrP in humans [27].
Strain theory is now applicable to most prion-like proteins (Figure 2)[28][29]. α-Synuclein, for example, has been known to be the culprit behind characteristically distinct pathologies, i.e., PD, DLB, and multiple system atrophy, while microtubule-associated protein tau is involved in multiple different tauopathies either as the primary cause or as a co-pathology [30][31]. In the case of Aβ, it has been known for several years that different proteoforms vary in their capability to form amyloids, seeding proficiencies, three-dimensional conformations, transport mechanisms and toxicities [32][33]. Each proteoform can adopt and propagate in multiple conformations [34]. These conformers do not only possess distinct biochemical signatures but also have different stabilities, distribution and morphology in the brain [35]. Moreover, accumulating evidence shows that many neurodegenerative proteinopathies can exist as rapidly progressive and other clinically distinct variants even though the underlying prion-like proteins and mechanisms are the same [36][37]. The capability of one protein to give rise to clinically distinct disorders and alter the progression of a disease has further complicated the characterization of neurodegenerative proteinopathies.
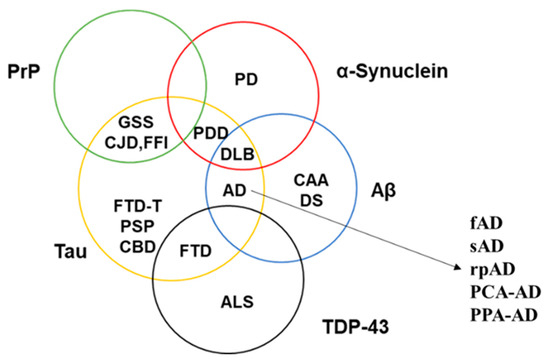
Figure 2. Involvement of known prion-like proteins in multiple neurodegenerative disorders. The figure depicts the overlapping pathological profile of PrP (green circle), α-Synuclein (red circle), Aβ (blue circle), Tau (yellow circle), and TDP-43 (black circle). Each of the stated disorders have further clinical variants (as shown in the case of AD), thereby complicating the role of prion-like proteins in bringing about the observed pathology. PDD—Parkinson’s disease with dementia; DS—Down’s syndrome; FTD-T—frontotemporal dementia with tau pathology; fAD—familial AD; sAD—sporadic AD; rpAD—rapidly-progressive AD; PCA-AD—posterior cortical atrophy–AD; PPA-AD—primary progressive aphasia with AD.
The study of prion-like proteins now encompasses the study of all variants/proteoforms rather than focusing on one parent entity. The existence of proteins as different functional variants is a known fact. These functional variants dictate the localization, uptake, recycling, and biological functions of a protein. In the case of prion-like proteins, the presence of distinct variants involved in neurodegenerative proteinopathies has been verified by several groups over the past two decades[12][14][38]. Although several different terms have been previously used in the literature to classify these variations, any prion-like protein can have:
-
Genetic variants (based on mutations).
-
Isoforms (based on differences in post-transcriptional modifications).
-
Proteoforms (based on differences in post-translation processing and three-dimensional conformation).
-
Strains (based on differences in infectivity and incubation periods).
With the acceptance of the notion that different isoforms, proteoforms, or strains of prion-like proteins may differ with respect to their molecular insult mechanisms and dictate the prognosis of associated pathology, the availability of high-resolution data about the sequence and structure has become the key in characterizing, diagnosing, and treating neurodegenerative proteinopathies [39][40][41][42][43][44]. It is therefore mandatory to establish tools that can provide insight into minor changes within the sequence, post-translational processing, and structure of a protein in its undigested form or native conformations.
References
- Alzheimer's Association; 2019 Alzheimer's disease facts and figures. Alzheimer's & Dementia 2019, 15, 321-387, 10.1016/j.jalz.2019.01.010.
- Prince, Martin; Maëlenn Guerchet; Matthew Prina.; The global impact of dementia 2013–2050.. Alzheimer's Disease International 2013, 8, 23.
- S.C. Johnson; T.W. Schmitz; C.H. Moritz; M.E. Meyerand; H.A. Rowley; A.L. Alexander; K.W. Hansen; C.E. Gleason; C.M. Carlsson; M.L. Ries; et al.S. AsthanaK. ChenE.M. ReimanG.E. Alexander Activation of brain regions vulnerable to Alzheimer's disease: The effect of mild cognitive impairment. Neurobiology of Aging 2006, 27, 1604-1612, 10.1016/j.neurobiolaging.2005.09.017.
- Qiying Sun; Nina Xie; Beisha Tang; Rena Li; Yong Shen; Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Frontiers in Molecular Neuroscience 2017, 10, 319, 10.3389/fnmol.2017.00319.
- Ramón Cacabelos; Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. International Journal of Molecular Sciences 2017, 18, 551, 10.3390/ijms18030551.
- Marc Manix; Piyush Kalakoti; Miriam Henry; Jai Deep Thakur; Richard Menger; Bharat Guthikonda; Anil Nanda; Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurgical Focus 2015, 39, E2, 10.3171/2015.8.focus15328.
- Johannes Levin; Alexander Kurz; Thomas Arzberger; Armin Giese; Günter U. Höglinger; The Differential Diagnosis and Treatment of Atypical Parkinsonism. Deutsches Aerzteblatt Online 2016, 113, 61-9, 10.3238/arztebl.2016.0061.
- Lokesh LokeshC Wijesekera; P Nigel Leigh; Amyotrophic lateral sclerosis. Orphanet Journal of Rare Diseases 2009, 4, 3-22, 10.1186/1750-1172-4-3.
- Vittorio Govoni; Elena Della Coletta; Edward Cesnik; Ilaria Casetta; Enrico Granieri; Can the age at onset give a clue to the pathogenesis of ALS?. Acta Neurologica Belgica 2016, 117, 221-227, 10.1007/s13760-016-0704-4.
- M. F. Knauer; B. Soreghan; D. Burdick; J. Kosmoski; C. G. Glabe; Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein.. Proceedings of the National Academy of Sciences 1992, 89, 7437-7441, 10.1073/pnas.89.16.7437.
- Gaël Nicolas; David Wallon; Claudia Goupil; Anne-Claire Richard; Cyril Pottier; Véronique Dorval; Mariana Sarov-Rivière; Florence Riant; Dominique Hervé; Philippe Amouyel; et al.Maelenn GuerchetBebene Ndamba-BandzouziPascal M'belessoJean-François DartiguesJean-Charles LambertPierre-Marie PreuxThierry FrebourgDominique CampionDidier HannequinElisabeth Tournier-LasserveSébastien S HébertAnne Rovelet-Lecrux Mutation in the 3’untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. European Journal of Human Genetics 2015, 24, 92-98, 10.1038/ejhg.2015.61.
- Norelle C. Wildburger; Thomas J. Esparza; Richard D. LeDuc; Ryan T. Fellers; Paul M. Thomas; Nigel J. Cairns; Neil L. Kelleher; Randall J. Bateman; David L. Brody; Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Scientific Reports 2017, 7, 1-9, 10.1038/s41598-017-10422-x.
- John Q Trojanowski; Michel Goedert; Takeshi Iwatsubo; Virginia M-Y Lee; Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death & Differentiation 1998, 5, 832-837, 10.1038/sj.cdd.4400432.
- John F. Kellie; Richard E. Higgs; John W. Ryder; Anthony Major; Thomas G. Beach; Charles H. Adler; Kalpana Merchant; Michael D. Knierman; Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson's Disease Brain Tissue by Intact Protein Mass Spectrometry. Scientific Reports 2014, 4, 5797, 10.1038/srep05797.
- Stanley B. Prusiner; D Westaway; Current ReviewInfectious and Genetic Manifestations of Prion Diseases. Molecular Plant-Microbe Interactions® 1991, 4, 226-33, 10.1094/mpmi-4-226.
- Zafar S.; Younas N.; Shafiq M.; Zerr I. . Prions—Some Physiological and Pathophysiological Aspects; IntechOpen: London, UK, 2018; pp. 362.
- Miguel Mompeán; Marco Baralle; Emanuele Buratti; Douglas Vinson Laurents; An Amyloid-Like Pathological Conformation of TDP-43 Is Stabilized by Hypercooperative Hydrogen Bonds. Frontiers in Molecular Neuroscience 2016, 9, 125, 10.3389/fnmol.2016.00125.
- Michael DiDonato; Lisa Craig; Mary E. Huff; Maria M. Thayer; Rosa M.F. Cardoso; Carey J. Kassmann; Terence P. Lo; Cami K. Bruns; Evan T. Powers; Jeffery W. Kelly; et al.Elizabeth D. GetzoffJohn A. Tainer ALS Mutants of Human Superoxide Dismutase Form Fibrous Aggregates Via Framework Destabilization. Journal of Molecular Biology 2003, 332, 601-615, 10.1016/s0022-2836(03)00889-1.
- Kathleen S. Molnar; N. Murat Karabacak; Joshua L. Johnson; Qi Wang; Ashutosh Tiwari; Lawrence J. Hayward; Stephen J. Coales; Yoshitomo Hamuro; Jeffrey N. Agar; A Common Property of Amyotrophic Lateral Sclerosis-associated Variants. Journal of Biological Chemistry 2009, 284, 30965-30973, 10.1074/jbc.m109.023945.
- Michel Goedert; Ross Jakes; Mutations causing neurodegenerative tauopathies. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2005, 1739, 240-250, 10.1016/j.bbadis.2004.08.007.
- SeoYoung Park; Jung Hoon Lee; Jun Hyoung Jeon; Min Jae Lee; Degradation or aggregation: the ramifications of post-translational modifications on tau. BMB Reports 2018, 51, 265-273, 10.5483/bmbrep.2018.51.6.077.
- Maria João Mascarenhas Saraiva; Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Human Mutation 2001, 17, 493-503, 10.1002/humu.1132.
- Laura Pont; Kader Poturcu; Fernando Benavente; José Barbosa; Victoria Sanz-Nebot; Comparison of capillary electrophoresis and capillary liquid chromatography coupled to mass spectrometry for the analysis of transthyretin in human serum. Journal of Chromatography A 2016, 1444, 145-153, 10.1016/j.chroma.2016.03.052.
- Marian DiFiglia; Ellen Sapp; Kathryn O. Chase; Stephen W. Davies; Gillian P. Bates; J. P. Vonsattel; Neil Aronin; Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990-1993, 10.1126/science.277.5334.1990.
- Dagmar E. Ehrnhoefer; Liza Sutton; Michael R. Hayden; Small Changes, Big Impact. The Neuroscientist 2011, 17, 475-492, 10.1177/1073858410390378.
- Rodrigo Morales; Prion strains in mammals: Different conformations leading to disease. PLOS Pathogens 2017, 13, e1006323-e1006323, 10.1371/journal.ppat.1006323.
- Patrick A. Lewis; M. Howard Tattum; Samantha Jones; Daljit Bhelt; Mark Batchelor; Anthony R. Clarke; John Collinge; Graham S. Jackson; Codon 129 polymorphism of the human prion protein influences the kinetics of amyloid formation. Journal of General Virology 2006, 87, 2443-2449, 10.1099/vir.0.81630-0.
- Cathrine Petersen, Amber L Nolan, Elisa de Paula França Resende, Zachary Miller, Alexander J Ehrenberg, Maria Luisa Gorno-Tempini, Howard J Rosen, Joel H Kramer, Salvatore Spina, Gil D Rabinovici, Bruce L Miller, William W Seeley, Helmut Heinsen, Lea Tenenholz Grinberg; Transmissible human proteopathies: an expanding field. Diagnostic Histopathology 2019, 25(1), 16-22, doi.org/10.1016/j.mpdhp.2018.11.002.
- Cathrine Petersen; Amber L. Nolan; Elisa De Paula França Resende; Zachary Miller; Alexander J. Ehrenberg; Maria Luisa Gorno-Tempini; Howard J. Rosen; Joel H. Kramer; Salvatore Spina; Gil D. Rabinovici; et al.Bruce L. MillerWilliam W. SeeleyHelmut HeinsenLea Tenenholz Grinberg Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathologica 2019, 138, 597-612, 10.1007/s00401-019-02036-6.
- Eduardo Tolosa; Jaume Campdelacreu; Clinical overview of the synucleinopathies. Movement Disorders 2003, 18, 21-27, 10.1002/mds.10559.
- David J. Irwin; Tauopathies as clinicopathological entities. Parkinsonism & Related Disorders 2016, 22, 29-33, 10.1016/j.parkreldis.2015.09.020.
- D Burdick; B Soreghan; M Kwon; J Kosmoski; M Knauer; A Henschen; J Yates; C Cotman; C Glabe; Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs.. Journal of Biological Chemistry 1992, 267, 546.
- Cynthia L. Martel; Jasmina B. Mackic; J.Gordon McComb; Jorge Ghiso; Berislav V. Zlokovic; Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer's amyloid β in guinea pigs. Neuroscience Letters 1996, 206, 157-160, 10.1016/s0304-3940(96)12462-9.
- Srirupa Chakraborty; Payel Das; Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Scientific Reports 2017, 7, 9941, 10.1038/s41598-017-10212-5.
- Jay Rasmussen; Mathias Jucker; Lary C. Walker; Aβ seeds and prions: How close the fit?. Prion 2017, 11, 215-225, 10.1080/19336896.2017.1334029.
- Christian Schmidt; Katharina Redyk; Bettina Meissner; Lennart Krack; Nico Von Ahsen; Sigrun Roeber; Hans Kretzschmar; Inga Zerr; Clinical Features of Rapidly Progressive Alzheimer’s Disease. Dementia and Geriatric Cognitive Disorders 2010, 29, 371-378, 10.1159/000278692.
- Paula Garcia-Esparcia; Georgios Sideris-Lampretsas; Karina Hernandez-Ortega; Oriol Grau-Rivera; Theodoros Sklaviadis; Ellen Gelpi; Isidro Ferrer; Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. American journal of neurodegenerative disease 2017, 6, 15-25.
- Marisol Espinoza; Rohan De Silva; Dennis W. Dickson; Peter Davies; Differential Incorporation of Tau Isoforms in Alzheimer's Disease. Journal of Alzheimer's Disease 2008, 14, 1-16, 10.3233/jad-2008-14101.
- Marina Boban; Helena Šarac; Ninoslav Mimica; Mihovil Mladinov; Christine Süßmair; Nibal Ackl; Benedikt Bader; Miljenko Huzak; Adrian Danek; Patrick R. Hof; et al.Goran Šimić CSF tau proteins in differential diagnosis of dementia. Translational Neuroscience 2010, 1, 43-48, 10.2478/v10134-010-0013-z.
- Denise Galante; Alessandro Corsaro; Tullio Florio; Serena Vella; Aldo Pagano; Francesca Sbrana; Massimo Vassalli; Angelo Perico; Cristina D’Arrigo; Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1-42 and Aβpy3-42 beta-amyloids. The International Journal of Biochemistry & Cell Biology 2012, 44, 2085-2093, 10.1016/j.biocel.2012.08.010.
- Kevin C. Stein; Heather L. True; Prion Strains and Amyloid Polymorphism Influence Phenotypic Variation. PLOS Pathogens 2014, 10, e1004328, 10.1371/journal.ppat.1004328.
- Christian Peters; Denisse Bascuñán; Carlos Opazo; Luis G. Aguayo; Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. Journal of Alzheimer's Disease 2016, 51, 689-699, 10.3233/jad-150896.
- Wei Qiang; Wai-Ming Yau; Jun-Xia Lu; John Collinge; Wei Qiang Wai-Ming Yau Jun-Xia Lu Robert Tycko; Structural variation in amyloid-β fibrils from Alzheimer's disease clinical subtypes. Nature 2017, 541, 217-221, 10.1038/nature20814.
- Diego Grassi; Shannon Howard; Minghai Zhou; Natalia Diaz-Perez; Nicolai T. Urban; Debbie Guerrero-Given; Naomi Kamasawa; Laura A. Volpicelli-Daley; Philip Lograsso; Corinne Ida Lasmézas; et al. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proceedings of the National Academy of Sciences 2018, 115, 2634-2643, 10.1073/pnas.1713849115.

