 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wei-Zheng Zhang | + 1804 word(s) | 1804 | 2021-03-15 09:13:36 | | | |
| 2 | Lily Guo | Meta information modification | 1804 | 2021-03-22 05:28:18 | | |
Video Upload Options
Hyperuricemia is a risk factor for gout. It has been well observed that a large proportion of individuals with hyperuricemia have never had a gout flare(s), while some patients with gout can have a normuricemia. This raises a puzzle of the real role of serum uric acid (SUA) in the occurrence of gout flares. As the molecule of uric acid has its dual effects in vivo with antioxidant properties as well as being an inflammatory promoter, it has been placed in a delicate position in balancing metabolisms. Gout seems to be a multifactorial metabolic disease and its pathogenesis should not rely solely on hyperuricemia or monosodium urate (MSU) crystals.
1. Introduction
Hyperuricemia has been defined as serum uric acid (SUA) >6.0 mg/dL in women; >7.0 mg/dL in men; >5.5 mg/dL in children and adolescents [1], and is an independent risk factor of a strong non-linear concentration-dependent to the incident of gout [2]. Genetic variants contribute largely to hyperuricemia [3] with 43 genes so far that have been identified in controlling SUA levels [4]. Gout, a common metabolic disorder with symptoms of localized inflammation, is caused by chronic and/or episodic deposition of monosodium urate (MSU) crystals in joints and soft tissues prompting a gouty attack/flare [5]. It has not been viewed just as an articular disease with its broad definition, but as a complex disease with interactive mechanisms of inflammatory and metabolic disorders displaying its symptoms beyond the local inflammatory consequence of MSU crystal deposition. Approximately up to 10% of adults have reported having gout [6], and 3.9% and 14.6% of the US population have gout and hyperuricemia, respectively [7].
It has been well observed and puzzled for a long time that not all hyperuricemia patients suffer from gout [8], only up to 36% of the patients develop gout [9][10], and not all gout patients have hyperuricemia [11][12]. Up to 76% of asymptomatic hyperuricemia patients could not find MSU crystal deposition [10]. This discrepancy has raised many clinical and scientific debates on the role of SUA in the development of gout and the efficacy of uric acid-lowering therapy (ULT) on the treatments of gout and other pathological conditions, despite the risk of gout increasing dramatically with increasing SUA levels in conjunction with additional factors [13]. The typical progression of gout could start from hyperuricemia without MSU crystal deposition, to crystal deposition without symptomatic gout, to acute gout flares and tophi, and to chronic gouty arthritis [14], but the correlations between gout and hyperuricemia are loose. Recent studies postulate that gout formation would be well beyond MSU crystal deposition with pathogenic mechanisms involving overproduction of chemotactic cytokines, cell proliferation and inflammation [15], and internalization of SUA induced pro-apoptotic and inflammatory effects [16].
Treatment of hyperuricemia in individuals without gout has been contentious partially due to the interrelationship between hyperuricemia and gout being not fully understood. Exploring such a mechanism, therefore, would be helpful in guiding the establishment of proper therapeutic regimes for both. This critical review aims to gather recently published literature for the elucidation of the interrelationship between hyperuricemia and gout in their pathogenesis, clinical evidence of the inflammation effects, MSU crystal formation, and therapeutic regimes, and attempts to explain why hyperuricemia does not necessarily induce gout.
2. Uric Acid and Gout
Uric acid (UA) is the end catabolic product of exogenous and endogenous purine nucleotide metabolism in humans. It exists in blood serum/plasma, cells, and tissues with steady-state conditions of its production and disposal. Its production can be found in almost all tissues, while its major disposal is via kidneys. SUA concentrations can be reflected by the intake from diet, in vivo purine metabolism, renal secretion, and intestinal degeneration [17]. In vivo, it may offer a neuroprotective advantage in the neurodegenerative Alzheimer’s disease [18], schizophrenia [19], Parkinson’s disease [20], multiple sclerosis [21], and serves as a depression biomarker [22]. SUA concentrations are linked to muscle strength and lean mass [23], although this was not shown in gastrointestinal tract cancer patients [24]. SUA may serve as a risk factor to predict poor thyroid function [25] or an indicator of malnutrition [15]. At a higher level, it activates inflammatory and oxidative mechanism action events in healthy subjects [26] and is a protective factor against the pathological decline of lung function [27] or an independent predictor for non-alcoholic fatty liver disease [28]. Abnormal SUA levels, either higher or lower, could increase the risk for mortality [29][30]. However, controversial reports have been presented: a slight increase in SUA level was an independent risk factor for all-cause and cardiovascular mortality [31][32], while another report did not find any relationship between SUA and cardiovascular disease (CVD) mortality and morbidity [33]. UA is also a potent antioxidant and an effective scavenger of singlet oxygen and free radicals [34], almost tenfold greater than other antioxidants in blood [26] or accounting for over half of the free radical scavenging activity in vivo [35]. Supplement of UA in donor blood sustains the antioxidant protection of the stored red blood cells [36]. The oxidant-antioxidant paradox of UA [37] may suggest UA could have different molecular behaviors under various pathological conditions. At the hydrophilic condition, it shows the protective effects of antioxidants [38]. Reducing SUA could decline its protective effect to radiation damage [39], and total bone mineral density [40][41] or a protective effect on bone loss in rheumatoid arthritis [42] (Figure 1).
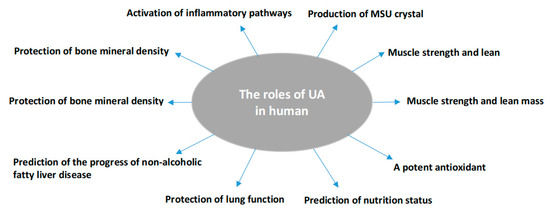
Figure 1. Summary of the pathophysiologic roles of uric acid in humans as identified in the review.
Beyond its role in protection, over-saturated SUA together with sodium could deposit in joints, soft tissues, bones, skin, etc., as MSU crystals to form tophi and trigger gout flares with episodes of severe pain. Gout is a common and complex form of arthritis with a sudden attack(s) of pain, swelling, redness, and tenderness in the affected location(s). Tophaceous gout has been defined as classic periarticular subcutaneous tophi, disseminated intradermal tophi, an ulcerative form, and gouty panniculitis [43] and commonly appears as firm, pink nodules or fusiform swellings [44]. Without clinical intervention, tophi can become developed within affected joints and or tissues and progressively damage them. Interestingly, the prevalence of gout flares, irrespective of SUA levels, has been linked to mental disorders [45][46]. Chronic heart failure and diabetes mellitus are more strongly associated with increased MSU crystal deposition in knees and feet/ankles than gout duration [47]. As reducing SUA may not be the only way to eliminate gout flares [48], the level of SUA, in essence, should be an indicator of oxidative paradox in vivo.
3. Factors Affecting MSU Crystal Deposition Other Than Hyperuricemia
It has been found that only about half of those with SUA concentrations of ≥10 mg/dL developed clinically evident gout during a 15-year period [2]. Despite hyperuricemia playing a critical role in the formation of MSU crystals, other factors affecting MSU crystallization in tissues to induce gout flare are also involved including but not limited to temperature, pH, ion concentrations, proteins, and various connective tissue conditions as well as secondary nucleation formation [49]. Additionally, white blood cell count (WBC) in synovial envirofluid is also significantly associated with the formation of MSU crystals [50], even though it is still been unresolved whether WBC induces MSU crystal formation or vice versa. Reducing blood lipid levels with a lipid-lowering agent(s) could concurrently reduce SUA level [51], suggesting a decline of the hydrophobic environment could enhance the solubility of SUA to be easily excreted by the kidney instead of forming MSU. Furthermore, hyperlipidemia is more commonly seen in patients with gout in comparison with asymptomatic individuals with HDL-C being a protective predictor of SUA levels [52].
In spite of hyperuricemia, it has been established that pre-biological fiber damage could be a prerequisite for MSU crystal deposition [53]. In the predominated crystal-rich areas of gouty tophi, new crystals add on the already formed crystals to form secondary nucleation which puts pressure on surrounding cells and causes tissue damage [54]. Osteoarthritis could alter the cartilage surface to precede MSU crystal formation happening at collagen-rich sites of damaged and exposed tissues [55], and surgical tissue damage could also induce gout flare [56]. The altered composition of microbiomes could then contribute to gout formation possibly due to stone or crystal growth in vivo [57]. All of that evidence may suggest that the formation of MSU crystals to induce a gout flare usually has an abnormal environment caused by a pathological condition(s) in addition to hyperuricemia.
4. The Causes of Hyperuricemia Irrelevant to Gout
An increase in UA concentration that exceeds the normal range might not be exclusively linked to gout flares [58] as many factors causing hyperuricemia are not relevant or significant to gout formation. Certain foods, status, or medicines could induce hyperuricemia. Under renal dis-function or cell damage, SUA could be suddenly increased by changing renal function to cause hyperuricemia [59]. Drugs such as diuretics (thiazide), anticonvulsants (valproate and phenobarbital), cyclosporine, theophylline, and pyrazinamide have been reported to increase SUA levels [60] in addition to favipiravir (an antiviral drug) [61]. Emotional stress [62], fasting [58], or dehydration [63] caused by physical activity can also increase the concentration of SUA. Although occasionally an acute gout flare may be linked with the medication(s) or condition(s), the enhanced SUA should be only temporarily sustained and a gout flare is considered unlikely to occur if a longer duration and higher dose treatment are avoided [64]. This may match with the fact that most gout patients do not know the trigger(s) of their gout flares [65]. Contrarily, a sudden reduction of SUA may also trigger a gout flare through the dissolution of MSU fallen off from tophi [66]. Allopurinol, a ULT medicine, has not shown any efficacy in the prevention of a first gout flare in patients with asymptomatic hyperuricemia [67]. Fundamentally, SUA level is associated with physical capacity and muscle strength in healthy subjects [23][24] and may only be a biological marker of non-gout conditions such as cardiovascular damage but is not a risk factor for its development [68]. Controversially, pseudo-gout, which is caused by calcium pyrophosphate deposition (CPPD), could have the same inflammatory symptoms as gout without hyperuricemia [69].
Gout can be self-resolvable with symptoms disappearing within days or weeks when hyperuricemia might still be sustained. Therefore, the symptoms might not be paralleled with a reduction in SUA level. The immunoreaction generated anti-UA antibody could also prevent or reduce inflammation conditions to release gout flare symptoms [70]. Under hyperuricemia, apoA-I elevation [71] or neutrophil-derived microvesicles [72] may play a role in the spontaneous resolution of acute gout arthritis.
The increased SUA after fasting for a long term may not induce any gout flares [58], plausibly as fasting could reduce the signaling pathway of the mammalian target of rapamycin (mTOR). The declined mTOR pathway could concomitantly inhibit cell growth, enhance autophagy and decrease activation of the NF-κB pathway as well as oxidative stress [73], thus reducing inflammatory processes and preventing a gout attack. All of those may be indicative that many elements inducing hyperuricemia could be irrelevant or insignificant to a gout flare. This may also at least partially explain the existence of asymptomatic hyperuricemia. However, longitudinal studies would be useful to understand the evolution of hyperuricemia and gout further and highlight the need for different treatment strategies [74].
References
- Kasper, D.; Hauser, S.; Longo, D.; Jameson, J.L.; Loscalzo, J. Harrison’s Principles of Internal Medicine, 19th ed.; Mcgraw-Hill: New York, NY, USA, 2015.
- Brucato, A.; Cianci, F.; Carnovale, C. Management of Hyperuricemia in Asymptomatic Patients: A Critical Appraisal. Eur. J. Intern. Med. 2020, 74, 8–17.
- Zhang, Q.; Gong, H.; Lin, C.; Liu, Q.; Baima, Y.; Wang, Y.; Lin, J. The prevalence of gout and hyperuricemia in middle-aged and elderly people in Tibet Autonomous Region, China: A preliminary study. Medicine 2020, 99, e18542.
- Boocock, J.; Leask, M.; Okada, Y.; Matsuo, H.; Kawamura, Y.; Shi, Y.; Li, C.; Mount, D.B.; Mandal, A.K.; Wang, W.; et al. Genomic dissection of 43 serum urate-associated loci provides multiple insights into molecular mechanisms of urate control. Hum. Mol. Genet. 2020, 29, 923–943.
- Towiwat, P.; Chhana, A.; Dalbeth, N. The Anatomical Pathology of Gout: A Systematic Literature Review. BMC Musculoskelet. Disord. 2019, 20, 140.
- Kuo, C.F.; Grainge, M.J.; Zhang, W.; Doherty, M. Global epidemiology of gout: Prevalence, incidence and risk factors. Nat. Rev. Rheumatol. 2015, 11, 649–662.
- Singh, G.; Lingala, B.; Mithal, A. Gout and hyperuricaemia in the USA: Prevalence and trends. Rheumatology 2019, 58, 2177–2180.
- Bardin, T.; Richette, P. Definition of hyperuricemia and gouty conditions. Curr. Opin. Rheumatol. 2014, 26, 186–191.
- Lin, K.C.; Lin, H.Y.; Chou, P. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. J. Rheumatol. 2000, 27, 1501–1505.
- Dalbeth, N.; House, M.E.; Aati, O.; Tan, P.; Franklin, C.; Horne, A.; Gamble, G.D.; Stamp, L.K.; Doyle, A.J.; McQueen, F.M. Urate crystal deposition in asymptomatic hyperuricaemia and symptomatic gout: A dual energy CT study. Ann. Rheum. Dis. 2015, 74, 908–911.
- Schlesinger, N.; Norquist, J.M.; Watson, D.J. Serum urate during acute gout. J. Rheumatol. 2009, 36, 1287–1289.
- Lee, J.S.; Kwon, O.C.; Oh, J.S.; Kim, Y.G.; Lee, C.K.; Yoo, B.; Hong, S. Clinical features and recurrent attack in gout patients according to serum urate levels during an acute attack. Korean J. Intern. Med. 2020, 35, 240–248.
- Singh, A.J.; Reddy, S.G.; Kundukulam, J. Risk factors for gout and prevention: A systematic review of the literature. Curr. Opin. Rheumatol. 2011, 23, 192–202.
- Dalbeth, N.; Stamp, L. Hyperuricaemia and gout: Time for a new staging system? Ann. Rheum. Dis. 2014, 73, 1598–1600.
- Bonino, B.; Leoncini, G.; Russo, E.; Pontremoli, R.; Viazzi, F. Uric acid in CKD: Has the jury come to the verdict? J. Nephrol. 2020, 33, 715–724.
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE 2014, 9, e115210.
- Rahimi-Sakak, F.; Maroofi, M.; Rahmani, J.; Bellissimo, N.; Hekmatdoost, A. Serum uric acid and risk of cardiovascular mortality: A systematic review and dose-response meta-analysis of cohort studies of over a million participants. BMC Cardiovasc. Disord. 2019, 19, 218.
- Spitsin, S.; Koprowski, H. Role of uric acid in Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1337–1338.
- He, Q.; You, Y.; Yu, L.; Yao, L.; Lu, H.; Zhou, X.; Wu, S.; Chen, L.; Chen, Y.; Zhao, X. Uric acid levels in subjects with schizophrenia: A systematic review and meta-analysis. Psychiatry Res. 2020, 292, 113305.
- Ellmore, T.M.; Suescun, J.; Castriotta, R.J.; Schiess, M.C. A Study of the Relationship Between Uric Acid and Substantia Nigra Brain Connectivity in Patients with REM Sleep Behavior Disorder and Parkinson’s Disease. Front. Neurol. 2020, 11, 815.
- Rentzos, M.; Nikolaou, C.; Anagnostouli, M.; Rombos, A.; Tsakanikas, K.; Economou, M.; Dimitrakopoulos, A.; Karouli, M.; Vassilopoulos, D. Serum uric acid and multiple sclerosis. Clin. Neurol. Neurosurg. 2006, 108, 527–531.
- Meng, X.; Huang, X.; Deng, W.; Li, J.; Li, T. Serum uric acid a depression biomarker. PLoS ONE 2020, 15, e0229626.
- Floriano, J.P.; Nahas, P.C.; de Branco, F.M.S.; Dos Reis, A.S.; Rossato, L.T.; Santos, H.O.; Limirio, L.S.; Ferreira-Filho, S.R.; de Oliveira, E.P. Serum Uric Acid Is Positively Associated with Muscle Mass and Strength, but Not with Functional Capacity, in Kidney Transplant Patients. Nutrients 2020, 12, 2390.
- Trindade, D.B.; de Araujo, V.A.; Franco, E.P.; Fernandes, R.C.; Carvalho, A.; Pimentel, G.D. Serum uric acid concentration is not associated with handgrip strength, lean body mass or survival in gastrointestinal cancer patients. Clin. Nutr. ESPEN 2020, 37, 75–79.
- Wang, X.J.; Qian, X.W.; Zhang, X.; Han, L.; Zheng, Y.Q.; Wu, T.; Qin, G.Y.; Ye, Z.B.; Xiao, J. Association of serum uric acid with thyroid function in health check-up participants. Chin. Med. J. 2020, 133, 1409–1414.
- Tariq, M.A.; Shamim, S.A.; Rana, K.F.; Saeed, A.; Malik, B.H. Serum Uric Acid—Risk Factor for Acute Ischemic Stroke and Poor Outcomes. Cureus 2019, 11, e6007.
- Fujikawa, H.; Sakamoto, Y.; Masuda, N.; Oniki, K.; Kamei, S.; Nohara, H.; Nakashima, R.; Maruta, K.; Kawakami, T.; Eto, Y.; et al. Higher Blood Uric Acid in Female Humans and Mice as a Protective Factor against Pathophysiological Decline of Lung Function. Antioxidants 2020, 9, 387.
- Wei, F.; Li, J.; Chen, C.; Zhang, K.; Cao, L.; Wang, X.; Ma, J.; Feng, S.; Li, W.D. Higher Serum Uric Acid Level Predicts Non-alcoholic Fatty Liver Disease: A 4-Year Prospective Cohort Study. Front. Endocrinol. 2020, 11, 179.
- Kuo, C.F.; See, L.C.; Yu, K.H.; Chou, I.J.; Chiou, M.J.; Luo, S.F. Significance of serum uric acid levels on the risk of all-cause and cardiovascular mortality. Rheumatology 2013, 52, 127–134.
- Seet, R.C.; Kasiman, K.; Gruber, J.; Tang, S.Y.; Wong, M.C.; Chang, H.M.; Chan, Y.H.; Halliwell, B.; Chen, C.P. Is uric acid protective or deleterious in acute ischemic stroke? A prospective cohort study. Atherosclerosis 2010, 209, 215–219.
- Konta, T.; Ichikawa, K.; Kawasaki, R.; Fujimoto, S.; Iseki, K.; Moriyama, T.; Yamagata, K.; Tsuruya, K.; Narita, I.; Kondo, M.; et al. Association between serum uric acid levels and mortality: A nationwide community-based cohort study. Sci. Rep. 2020, 10, 6066.
- Tai, S.; Li, X.; Zhu, Z.; Tang, L.; Yang, H.; Fu, L.; Hu, X.; Fang, Z.; Zhou, S. Hyperuricemia is a Risk Factor for One-Year Overall Survival in Elderly Female Patients with Acute Coronary Syndrome. Cardiovasc. Ther. 2020, 2020, 2615147.
- Sieminska, E.; Sobczak, P.; Skibinska, N.; Sikora, J. The differential role of uric acid—The purpose or cause of cardiovascular diseases? Med. Hypotheses 2020, 142, 109791.
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862.
- Bowman, G.L.; Shannon, J.; Frei, B.; Kaye, J.A.; Quinn, J.F. Uric acid as a CNS antioxidant. J. Alzheimers Dis. 2010, 19, 1331–1336.
- Bardyn, M.; Chen, J.; Dussiot, M.; Crettaz, D.; Schmid, L.; Langst, E.; Amireault, P.; Tissot, J.D.; Jolicoeur, M.; Prudent, M. Restoration of Physiological Levels of Uric Acid and Ascorbic Acid Reroutes the Metabolism of Stored Red Blood Cells. Metabolites 2020, 10, 226.
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619.
- Stewart, J.D.; Langlois, V.; Noone, D. Hyperuricemia and Hypertension: Links and Risks. Integr. Blood Press. Control 2019, 12, 43–62.
- Paithankar, J.G.; Kudva, A.K.; Raghu, S.V.; Patil, R.K. Radioprotective role of uric acid: Evidence from studies in Drosophila and human dermal fibroblast cells. Mol. Biol. Rep. 2020, 47, 2427–2436.
- Pan, K.; Yao, X.; Liu, M.; Zhu, Z. Association of Serum Uric Acid Status with Bone Mineral Density in Adolescents Aged 12–19 Years. Front. Med. 2020, 7, 255.
- Yao, X.; Chen, L.; Xu, H.; Zhu, Z. The Association between Serum Uric Acid and Bone Mineral Density in Older Adults. Int. J. Endocrinol. 2020, 2020, 3082318.
- Lee, H.N.; Kim, A.; Kim, Y.; Kim, G.T.; Sohn, D.H.; Lee, S.G. Higher serum uric acid levels are associated with reduced risk of hip osteoporosis in postmenopausal women with rheumatoid arthritis. Medicine 2020, 99, e20633.
- Aguayo, R.S.; Baradad, M.; Soria, X.; Abal, L.; Sanmartin, V.; Egido, R.; Gallel, P.; Casanova, J.M.; Marti, R.M. Unilateral milia-type intradermal tophi associated with underlying urate subcutaneous deposition: An uncommon cutaneous presentation of gout. Clin. Exp. Dermatol. 2013, 38, 622–625.
- Pradhan, S.; Sinha, R.; Sharma, P.; Sinha, U. Atypical Cutaneous Presentation of Chronic Tophaceous Gout: A Case Report. Indian Dermatol. Online J. 2020, 11, 235–238.
- Howren, A.; Bowie, D.; Choi, H.K.; Rai, S.K.; De Vera, M.A. Epidemiology of depression and anxiety in gout: A systematic review and metaanalysis. J. Rheumatol. 2020, 48, 129–137.
- Zhou, Q.; Shao, Y.C.; Gan, Z.Q.; Fang, L.S. Lower vitamin D levels are associated with depression in patients with gout. Neuropsychiatr. Dis. Treat. 2019, 15, 227–231.
- Pascart, T.; Ramon, A.; Ottaviani, S.; Legrand, J.; Ducoulombier, V.; Houvenagel, E.; Norberciak, L.; Richette, P.; Becce, F.; Ornetti, P. Association of Specific Comorbidities with Monosodium Urate Crystal Deposition in Urate-Lowering Therapy-Naive Gout Patients: A Cross-Sectional Dual-Energy Computed Tomography Study. J. Clin. Med. 2020, 9, 1295.
- Sun, R.; Lu, J.; Li, H.; Cheng, X.; Xin, Y.; Li, C. Evaluation of Febuxostat Initiation During an Acute Gout Attack: A Prospective, Randomized Clinical Trial. Jt. Bone Spine 2020, 87, 461–466.
- Chhana, A.; Lee, G.; Dalbeth, N. Factors influencing the crystallization of monosodium urate: A systematic literature review. BMC Musculoskelet. Disord. 2015, 16, 296.
- Aliste-Fernandez, M.; San-Jose, P.; Aguadero, V. White blood cell count and total protein concentration to predict the absence of microcrystals in synovial fluid. Clin. Biochem. 2020, 83, 81–85.
- Cao, J.Y.; Waldman, B.; O’Connell, R.; Sullivan, D.R.; Scott, R.S.; Aryal, N.; Gebski, V.; Marschner, I.; Taskinen, M.R.; Simes, J.R.; et al. Uric acid predicts long-term cardiovascular risk in type 2 diabetes but does not mediate the benefits of fenofibrate: The FIELD study. Diabetes Obes. Metab. 2020, 22, 1388–1396.
- Liang, J.; Jiang, Y.; Huang, Y.; Song, W.; Li, X.; Ou, J.; Wei, Q.; Gu, J. The comparison of dyslipidemia and serum uric acid in patients with gout and asymptomatic hyperuricemia: A cross-sectional study. Lipids Health Dis. 2020, 19, 31.
- Pascual, E.; Addadi, L.; Andres, M.; Sivera, F. Mechanisms of crystal formation in gout-a structural approach. Nat. Rev. Rheumatol. 2015, 11, 725–730.
- Martillo, M.A.; Nazzal, L.; Crittenden, D.B. The crystallization of monosodium urate. Curr. Rheumatol. Rep. 2014, 16, 400.
- Pritzker, K.P.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29.
- Dang, N.L.H.; Kim, J.K.; Lee, K.B. Crystal-Induced Arthritis After Total Ankle Arthroplasty. J. Am. Podiatr. Med. Assoc. 2019, 109, 159–161.
- Ning, Y.; Yang, G.; Chen, Y.; Zhao, X.; Qian, H.; Liu, Y.; Chen, S.; Shi, G. Characteristics of the Urinary Microbiome from Patients With Gout: A Prospective Study. Front. Endocrinol. 2020, 11, 272.
- Wilhelmi de Toledo, F.; Grundler, F.; Goutzourelas, N.; Tekos, F.; Vassi, E.; Mesnage, R.; Kouretas, D. Influence of Long-Term Fasting on Blood Redox Status in Humans. Antioxidants 2020, 9, 496.
- Yamamoto, T.; Xie, J.; Li, Z.; Field, C.; Block, C.; Taylor, T. Effect of Uric Acid Control on Serum Creatinine. J. Clin. Rheumatol. 2019, 25, 279–283.
- Kubota, M. Hyperuricemia in Children and Adolescents: Present Knowledge and Future Directions. J. Nutr. Metab. 2019, 2019, 3480718.
- Mishima, E.; Anzai, N.; Miyazaki, M.; Ab. Uric Acid Elevation by Favipiravir, an Antiviral Drug. Tohoku J. Exp. Med. 2020, 251, 87–90.
- Batandier, C.; Poyot, T.; Marissal-Arvy, N.; Couturier, K.; Canini, F.; Roussel, A.M.; Hininger-Favier, I. Acute emotional stress and high fat/high fructose diet modulate brain oxidative damage through NrF2 and uric acid in rats. Nutr. Res. 2020, 79, 23–34.
- Yoo, I.H.; Kim, W.; Cho, J.; Kim, H.; Lim, B.C.; Hwang, H.; Chae, J.H.; Choi, J.; Kim, K.J. Erratum to: Elevated Serum Uric Acid in Benign Convulsions with Mild Gastroenteritis in Children. J. Clin. Neurol. 2020, 16, 181.
- Hase, R.; Kurata, R.; Ishida, K.; Kurita, T.; Muranaka, E.; Mito, H. Acute Gouty Arthritis During Favipiravir Treatment for Coronavirus Disease 2019: A Case Report. Intern. Med. 2020, 59, 2327–2329.
- Abhishek, A.; Valdes, A.M.; Jenkins, W.; Zhang, W.; Doherty, M. Triggers of acute attacks of gout, does age of gout onset matter? A primary care based cross-sectional study. PLoS ONE 2017, 12, e0186096.
- Flynn, T.J.; Cadzow, M.; Dalbeth, N.; Jones, P.B.; Stamp, L.K.; Hindmarsh, J.H.; Todd, A.S.; Walker, R.J.; Topless, R.; Merriman, T.R. Positive association of tomato consumption with serum urate: Support for tomato consumption as an anecdotal trigger of gout flares. BMC Musculoskelet. Disord. 2015, 16, 196.
- Hazard, A.; Bourrion, B.; Dechaine, F.; Fournier, L.; Francois, M. Lack of evidence for allopurinol for the prevention of a first gout attack in asymptomatic hyperuricemia: A systematic review. Eur. J. Clin. Pharmacol. 2020, 76, 897–899.
- Cortese, F.; Giordano, P.; Scicchitano, P.; Faienza, M.F.; De Pergola, G.; Calculli, G.; Meliota, G. Uric acid in metabolic and cerebrovascular disorders: A review. Curr. Vasc. Pharmacol. 2019, 18, 610–618.
- Zamora, A.E.; Naik, R. Calcium Pyrophosphate Deposition Disease. Available online: (accessed on 31 December 2020).
- Felten, R.; Duret, P.M.; Gottenberg, J.E.; Spielmann, L.; Messer, L. At the crossroads of gout and psoriatic arthritis: “psout”. Clin. Rheumatol. 2020, 39, 1405–1413.
- Chiang, S.L.; Ou, T.T.; Wu, Y.J.; Tu, H.P.; Lu, C.Y.; Huang, C.M.; Kuo, T.M.; Wang, T.N.; Chou, C.H.; Ko, Y.C. Increased level of MSU crystal-bound protein apolipoprotein A-I in acute gouty arthritis. Scand. J. Rheumatol. 2014, 43, 498–502.
- Cumpelik, A.; Ankli, B.; Zecher, D.; Schifferli, J.A. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome. Ann. Rheum. Dis. 2016, 75, 1236–1245.
- Hwangbo, D.S.; Lee, H.Y.; Abozaid, L.S.; Min, K.J. Mechanisms of Lifespan Regulation by Calorie Restriction and Intermittent Fasting in Model Organisms. Nutrients 2020, 12, 1194.
- Filippou, G.; Scanu, A.; Adinolfi, A.; Picerno, V.; Toscano, C.; Bortoluzzi, A.; Frediani, B.; Govoni, M.; Punzi, L.; Scire, C.A. The two faces of the same medal... or maybe not? Comparing osteoarthritis and calcium pyrophosphate deposition disease: A laboratory and ultrasonographic study. Clin. Exp. Rheumatol. 2020, 39, 66–72.


