 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Małgorzata Jeleń | + 4375 word(s) | 4375 | 2021-03-10 08:02:07 | | | |
| 2 | Bruce Ren | + 7 word(s) | 4382 | 2021-03-16 12:24:27 | | | | |
| 3 | Bruce Ren | Meta information modification | 4382 | 2021-03-18 07:41:44 | | |
Video Upload Options
Azaphenothiazines are the largest and most perspective group of modified phenothiazines, and they exhibit variety of biological activities. The review sums up the current knowledge on the anticancer activity of isomeric pyridobenzothiazines and dipyridothiazines, which are modified azaphenothiazines with one and two pyridine rings, respectively, against 10 types of cancer cell lines. Some 10-substituted dipyridothiazines and even 10-unsubstituted parent compounds, such as 10H-1,9-diazaphenothiazine and 10H-3,6-diazaphenothiazine, exhibited very potent action with the IC50 values less than 1 µg/mL and 1 µM against selected cancer cell lines. The strength of the anticancer action depends both on the tricyclic ring scaffolds and the substituents at the thiazine nitrogen atom.
1. Introduction
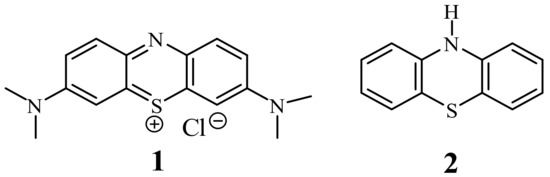
Phenothiazines are a very important class of heterocyclic compounds possessing a tricyclic dibenzo-[1,4]-thiazine ring system. This system, as the phenothiazinium salt, was for the first time synthesized by Caro in 1876 as a dye called methylene blue (1) [1]. Later in 1883, Bernthsen obtained free 10H-phenothiazine (2) in the reaction of sulfurization of diphenylamine with elemental sulfur [2] (Figure 1). In 1950s, the researchers at Rhône-Poulenc synthesized phenothiazines which exhibited antihistaminic and antipsychotic activities. Since this time, phenothiazines have been essential drugs (at least 100 phenothiazines) for the clinical treatment of psychotic disorders [3], among which chlorpromazine, thioridazine, fluphenazine, promethazine, and trifluoperazine are the most known. Those classical phenothiazines contained the dialkylaminoalkyl substituents at the thiazine nitrogen atom (in position 10) and additionally small substituents in position 2. They exhibit also analgesic, anti-emetic, antihelmentic, and antipuritic activities [3][4].
For a few decades, there has been an extremely high interest in searching for new derivatives of phenothiazines possessing different activities. The first approach is called drug repurposing or drug repositioning, and the use of known drugs for new biological targets decreases the cost of research, while maintaining quality and proven safety [5][6][7]. Phenothiazines with thioridazine at the lead are regarded as the most up and coming in a treatment of the multidrug resistant tuberculosis [8][9][10][11], cancer cells [5][6][7][12][13][14][15][16], and neurodegenerative diseases [13]. They also exhibit antiviral, anti-emetic, antipruritic, anti-inflammatory, antitussive, analgesic, antibacterial, and antifungal activities [13][14][17].
The second approach is based on the synthesis of new structurally modified phenothiazines. This modification involves the insertion of new substituents at the nitrogen atom, oxidation of the sulfide group into sulfoxide and sulfone groups in the thiazine ring, and the exchange of one or two benzene rings with other rings (homoaromatic and heteroaromatic). The last modification is the most perspective because it allows the formation of not only a new phenothiazine scaffold (not only tricyclic but also tetracyclic and pentacyclic), but also allows to introduce new substituents. The introduction of the azine ring such as pyridine, pyridazine, pyrimidine, pyrazine, or quinoline in the place of the benzene ring, leads to new derivatives of phenothiazines, azaphenothiazines. Over 50 types of azaphenothiazine scaffolds are known which belong to varied heterocyclic ring systems [18][19].
The anticancer activity of classical and modified phenothiazines was summarized for the first time by Motohashi in the book chapter in the late 1980s [20]. Later, he with his research groups started extensive studies on the anticancer activity of new phenothiazines which were the inspiration to other research groups. They synthesized modified tricyclic phenothiazines with amidoalkyl, sulfonamidoalkyl, and chloroethylureidoalkyl substituents in position 10 and replaced the benzene ring with naphthalene. These phenothiazines exhibited cytotoxic activities against ten different human tumor cell lines: leukemia, melanoma, small cell lung, colon, central nervous system, renal, breast, ovarian, and prostate tumors. Their results were published in over 50 original papers and reviews [21][22][23][24][25][26][27][28][29].
The modified phenothiazines exhibit not only anticancer activity, but a wide range of other activities such as the reversal of multidrug resistance, anti-inflammatory, antibacterial, antiviral, antiplasmid, cholinesterase inhibitory, antioxidant, and antihyperlipidemic and are considered potential agents in the treatment in Alzheimer’s and Creutzfeldt–Jakob diseases, published in hundreds of original papers and patents, and summarized in numerous comprehensive review papers and chapters [18][19][30][31][32][33][34][35][36][37][38].
The modification of the phenothiazine structures with the pyridine ring leads to different pyridobenzothiazines and dipyridothiazines, also named x-monoazaphenothiazines and x,y-diazaphenothiazines, the largest but also most diverse family of azaphenothiazines. The aim of this review is to present comprehensively anticancer activities of those azaphenothiazines.
2. Pyridobenzothiazines
Pyridobenzothiazines represent linearly condensed tricyclic ring systems where the 1,4-thiazine ring is fused with the benzene and pyridine rings along the C-C bonds. As they differ from classical phenothiazines in the presence of the additional nitrogen atom instead of the carbon atom, they are named azaphenothiazines (1-, 2-, 3- and 4-azaphenothiazines).
2.1. 1-Azaphenothiazines (Pyrido[3,2,-b]benzo[1,4]thiazines)
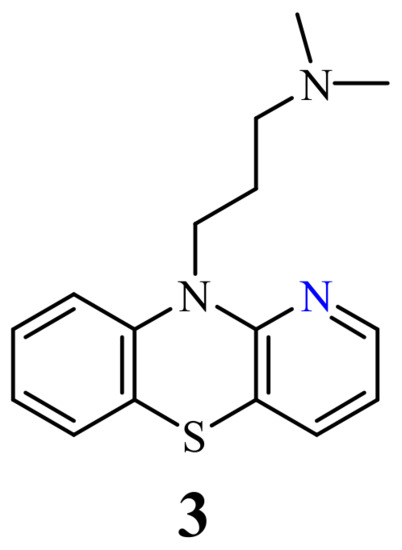
Of the 1-azaphenothiazines, antitumor activity has so far been described only for 10-substituted derivatives including a dimethylaminopropyl group (3), called prothipendyl (Figure 2), and a hexyl chain containing amino and nitro groups or a bromine atom at the end (4–9) (Figure 3).

The anticancer activity of prothipendyl was investigated recently using the cultured glioblastoma SNB-19, melanoma C-32, breast cancer MCF-7, and ductal carcinoma T47D cell lines with the normal human fibroblast (HFF-1) cell line used as a control. The cell lines used showed a different sensitivity to prothipendyl. The MCF-7 and the C32 cells lines were found to be the most sensitive with the IC50 value of 23.2 µg/mL and 28.1 µg/mL, respectively. The T47D and the SNB19 cells lines were the most resistant for the tested compound (with IC50 of 32.3 µg/mL and 36.6 µg/mL, respectively). Prothipendyl was found to be non-toxic (IC50 > 50 µg/mL) against the normal human fibroblast (HFF-1) cell line in comparison with cisplatin used as a reference (IC50 = 8.2 µg/mL) [45][46][47].
A set of twenty-one novel 10-substituted 1-azaphenothiazines (4–9) containing aminohexyl and bromohexyl groups together with a nitro group in the pyridine ring (Figure 3) were investigated for their action against six cancer cell lines (malignant brain cancer T98G, lung cancer H460, thyroid cancer SNU80, oral cancer KB, blood cancer THP1, and blood cancer U937) and two normal cell lines (the normal lung fibroblast MRC5 and the normal embryonic kidney cells HEK293) with the actinomycin D used as the standard drug in the studies.
Eleven of the compounds tested exhibited distinct inhibitory action against selected cancer lines (IC50 < 10 µg/mL) and were a few times more effective in comparison to a standard drug actinomycin D. Compound 6 (with the piperidinylhexyl group) showed the highest activity against the lung cancer H460, the malignant brain cancer T98G, and the thyroid cancer SNU80 with the IC50 values of 2.27–3.8 µg/mL, respectively. Similarly active were derivatives 9 (with the pyrrolidinyl-piperidinylhexyl group) against the H460 and SNU80 cell lines (IC50 = 2.1 and 2.3 µg/mL), 7 (morpholinylhexyl group), and 5 (pyrrolidinylhexyl) against H460 line (IC50 = 2.5 and 2.7 µg/mL). The derivative 4 was active against those three cell lines showing the IC50 values of 3.8–6.2 µg/mL [48].
Very recently, prothipendyl exhibited antiviral action against a mosquito-transmitting alphavirus (CHIKV) inducing CHIK fever [49][50].
Other derivatives of 1-azaphenothiazine exhibited mainly antihistaminic, anticholinergic, anti-emetic, antitussive, neuropharmacological, antibacterial, and antitubercular activities and were the subject of many publications and reviews [51][52][53][54][55][56][57][58][59][60][61][62].
2.2. 2-Azaphenothiazines (Pyrido[4,3-b]benzo[1,4]thiazines)
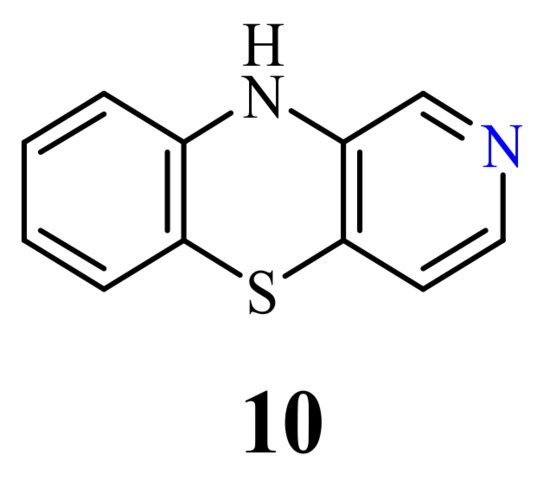
Despite the fact that 10H-2-azaphenothiazines (10) (Figure 4) was synthesized in the late 1950s [63][64], there are no reports in the literature about the antitumor activity of molecules containing the 2-azaphenothiazine skeleton. Instead of that, the aminoalkyl derivatives exhibited antipsychotic and sedative properties [65].
2.3. 3-Azaphenothiazines (Pyrido[3,4-b]benzo[1,4]thiazines)
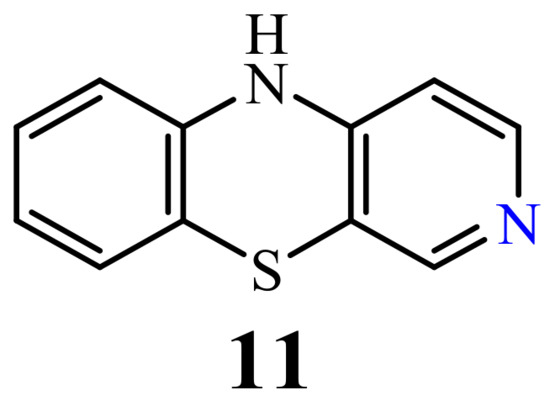
Although the synthesis of 10H-3-azaphenothiazine (11) (Figure 5) was described in the 1960s [66], antitumor activity has not yet been described but its derivatives exhibited sedative, hypnotic, anticonvulsant, and hypotensive activity [65][66]
2.4. 4-Azaphenothiazines (Pyrido[2,3-b]benzo[1,4]thiazines)
10H-4-Azaphenothiazine (12) (Figure 6) was obtained in 1958 [67]. Until now, only studies on their potential activity as allergy inhibitors were conducted for compounds containing the 4-azaphenothiazine skeleton [68].

As was discussed above, pyridobenzothiazines are not intensively explored nowadays, despite their wide range of biological activities.
The information provided on anticancer activity of pyridobenzothiazines was summarized in Table 1.
| Investigated Compounds | Substituents at the Thiazine Nitrogen Atom | Activity, IC50 [Ref] | Cancer Cell Lines |
|---|---|---|---|
| Pyridobenzothiazines | |||
| 1-Azaphenothiazine 3 |
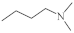 |
23.2 µg/mL [45][46][47] 28.1 µg/mL 32.3 µg/mL 36.6 µg/mL |
C-32 MCF-7 T47D SNB-19 |
| 4 |  |
3.8–6.2 µg/mL [48] | H460,T98G, SNU80 |
| 6 | 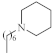 |
2.27–3.8 µg/mL [48] | H460, T98G, SNU80 |
| 7 | 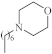 |
2.5 µg/mL [48] | H460 |
| 9 |  |
2.1–2.3 µg/mL [48] | H460, SNU80 |
| Dipyridothiazines | |||
| 1,6-Diazaphenothiazine 13 | H | 4.8 µg/mL [47] 7.5 µg/mL |
MCF-7 C-32 |
| 14 |  |
3.9 µg/mL [47] | MCF-7 |
| 15 | 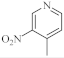 |
4.6 µg/mL [47] | MCF-7 |
| 16 | 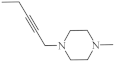 |
7.5 µg/mL [47] | MCF-7 |
| 17 | 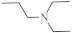 |
6.6 µg/mL [47] | C-32 |
| 18 |  |
18.9 µg/mL [47] | SNB-19 |
| 19 | 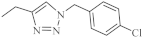 |
0.25-4.66 µM [69] | SNB-19, Caco-2, A549, MDA-MB231 |
| 1,8-Diazaphenothiazine | |||
| 20 | H | 5 µg/mL [70][71] 10 µg/mL |
SW-948 L-1210. |
| 23 | 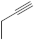 |
26-46 µg/mL [70] | SNB-19, C-32, T47D |
| 24 | 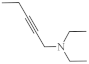 |
26.1 µg/mL [70] | C-32 |
| 25 |  |
1.82 µM [69] | A549 |
| 1,9-Diazaphenothiazine | |||
| 26 | H | 3.83 µM [72] | C-32 |
| 27 | 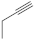 |
3.85 µM [72] 3.37 µM |
SNB-19 C-32 |
| 28 | 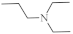 |
0.34 µM [72] 2.13 µM |
SNB-19 MDA-MB-321 |
| 2,7-Diazaphenothiazine | |||
| 29 | H | 0.3 µg/mL [73] 1.7 µg/mL 2.4 µg/mL 3.6 µg/mL 3.1 µg/mL 3.9 µg/mL 5.5 µg/mL 4.1 µg/mL 5.9 µg/mL 6.5 µg/mL 6.8 µg/mL 7.1 µg/mL 8.4 µg/mL |
HOP-62 HOP-92 colon 205 HTC-116 RXF 393 736-0 ACHN HL-60(TB) HS 578T M-14SF-539, SNB-19 OVCAR-8 PC-3 |
| 31 | 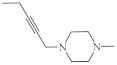 |
9.6 µg/mL [45] | T-47D |
| 32 | 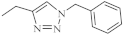 |
0.26 µM [69] 0.77 µM |
Caco-2, A549 MDA-MB231 |
| 33 | 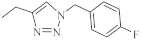 |
0.64 µM [69] 0.65 µM |
MDA-MB231 A549 |
| 3,6-Diazaphenothiazine | |||
| 34 | H | 0.46 µg/mL [46] 0.72 µg/mL 0.62 µM [74] |
SNB-19 C-32, MCF-7 A2780 |
| 35 | 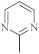 |
0.73 µg/mL [46] | MCF-7 |
| 36 |  |
6.3 µg/mL [46] 11.3 µg/mL |
C-32 MCF-7 |
| 37 |  |
0.11 µg/mL [75] | SNB-19 |
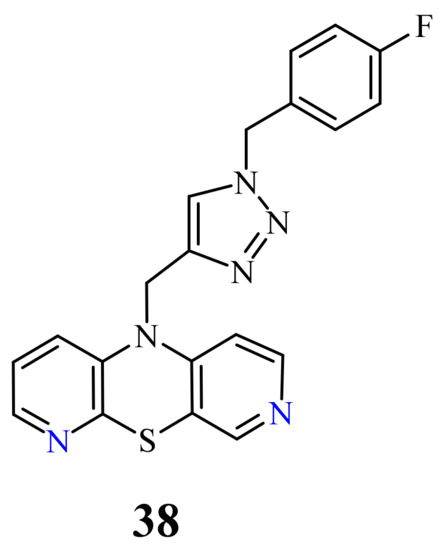
| 38 | 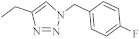 |
0.25 µM [69] | Caco-2, A549 |
3. Dipyridothiazines
Dipyridothiazines mean linearly condensed tricyclic ring systems where the 1,4-thiazine ring is fused with two pyridine rings along the C-C bonds. As they contain two additional nitrogen atoms (instead of two carbon atoms) in comparison with classical phenothiazines, they are named diazaphenothiazines. Out of ten theoretically possible dipyridothiazine systems, only six have been known as 1,6-, 1,8-, 1,9-, 2,7-, 3,6-, and 3,7-diazaphenothiazines. This small amount of well-known dipyridothiazine types is a result of the difficulties in their synthesis involving orto disubstituted (2,3- and 3,4-)pyridines [19].
3.1. 1,6-Diazaphenothiazines (Dipyrido[2,3-b;2’,3’-e][1,4]thiazines)
10H-1,6-Diazaphenothiazine (13) (Figure 7) was prepared in 1958 and was converted into a dimethylaminopropyl derivative. This compound exhibited slight synergic effect with known biomolecules such as morphine and barbiturates and weak action of lowering body temperature in relation to chlorpromazine [76].
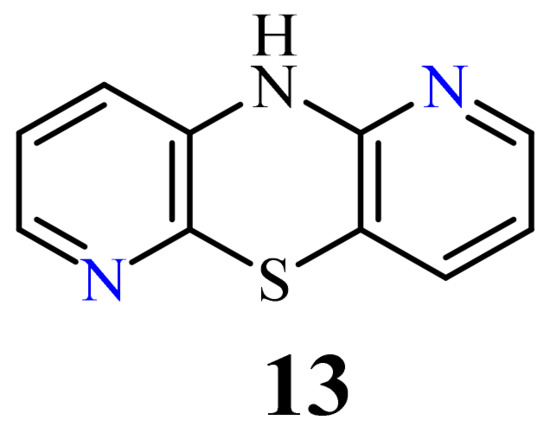
A large family of 1,6-Diazaphenothiazine derivatives (Figure 8) with the alkyl, heteroaryl, amidoalkyl, and dialkylaminoalkynyl groups was synthesized in 2016 and tested for their anticancer action against the glioblastoma SNB-19, melanoma C-32, and breast cancer MCF-7 cell lines [47]. The compounds exhibited diverse levels of activity (IC50 in the range of 3.9 to 49.1 µg/mL) depending on the type of substituent and the type of cell line. The parent compound, 10H-1,6-diazaphenothiazine (13), and compounds 14 and 15 (containing the propargyl and nitropyridinyl substituents at the thiazine nitrogen atom) were found to be more active (IC50 = 4.8, 3.9 and 4.6 µg/mL) in relation to cisplatin as a reference compound (7.4 µg/mL) against the breast cancer MCF-7 cells. The derivative 16 (containing the methylpiperazinylbutynyl substituent) was as effective as cisplatin (7.5 µg/mL). 10H-1,6-Diazaphenothiazine (13) and compound 17 (containing the diethylaminoethyl substituent) were slightly more effective (7.5 and 6.6 µg/mL) than cisplatin (7.8 µg/mL) against the C-32 cells. The derivative 18 (with the allyl substituent) was the most effective against the SNB-19 cells with the IC50 values of 18.9 µg/mL. The most anticancer active compounds were nontoxic with reference to normal fibroblasts HFF-1 with the IC50 values exceeded 50 µg/mL [47].
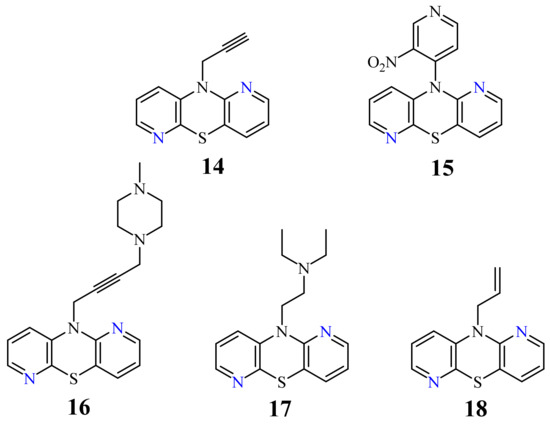
Further studies looking for the structure–activity relationship (SAR) related to lipophilicity analysis were performed; however, they did not show the absolute dependence of the anticancer activity on the lipophilic properties [77].
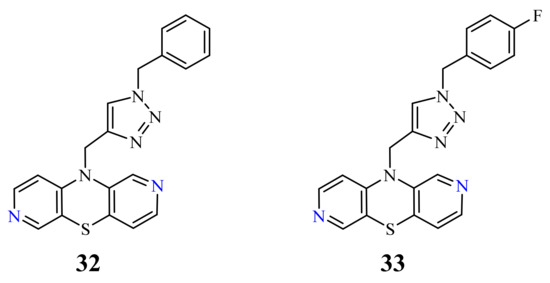
Recently, novel 1,2,3-triazole-dipyridothiazine hybrids containing the 1,6-diazaphenothiazine system and selected p-substituted benzyl substituents at the 1,2,3-triazole ring were obtained. The compound 19 (Figure 9) with the p-chlorobenzyl substituent showed an extremely promising anticancer activity (IC50 = 0.25–4.66 µM) in relation to the glioblastoma SNB-19, the colorectal carcinoma Caco-2, the lung cancer A549, and the breast cancer MDA-MB231 [69].
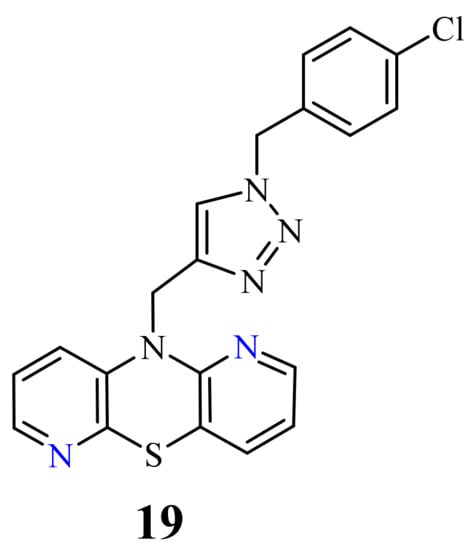
The compound was nontoxic against the normal fibroblasts NHDF with IC50 > 100 µM. This derivative was selected to study the mechanism of anticancer activity using RT-qPCR method. The influence on the gene transcriptional activities of proliferation marker (H3), the cell cycle regulator (TP53 and CDKNIA), and the intracellular apoptosis pathway (BCL-2 and BAX) were analyzed. The compound greatly affected the mRNA copy number of histone H3 gene in all cancer lines, which had an influence on a modification of the chromatin structure in the cells and significantly interfered with the amount of mRNA copies of the TP53 in all investigated cancer lines. There was also a strong decrease in the CDKN1A copies in all cancer cells, which can suggest a possibility of participation in cell cycle arrest. The derivative reduced remarkably the expression of the BCL-2 in SNB-19, A549, and MDA-MB231 cancer lines, but in the Caco-2 cell line there was a slight increase. The analysis of expression of H3, TP53, CDKN1A, BCL-2, and BAX genes revealed that this compound inhibited the proliferation in all cells and activated mitochondrial events of apoptosis [69].
3.2. 1,8-Diazaphenothiazines (Dipyrido[2,3-b;3’,4’-e][1,4]thiazines)
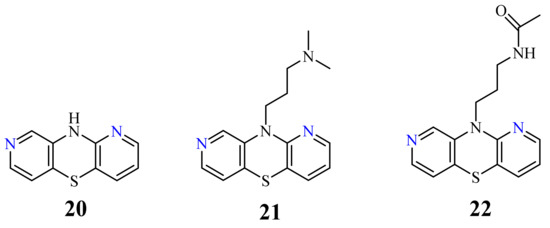
A group of 1,8-diazaphenothiazines was first reported in 2015. The biological potential of the compounds was the subject of both the publication and the patent [70][71]. 10H-1,8-Diazaphenothiazine 20 (Figure 10) and its 10-substituted derivatives containing various groups, such as the alkyl, heteroaryl, dialkylaminoalkyl, amidoalkyl, and sulfonamidoalkyl, were tested for their pharmacological activities. The test of proliferative response of the human peripheral blood mononuclear cells (PBMC) induced by the phytohemagglutinin A (PHA) exhibited a meaningful action of compound 20 and the derivative 21 (containing the dimethylaminopropyl substituent, Figure 10) at concentration of 50 µg/mL. These compounds showed the strongest inhibition of the tumor necrosis alpha (TNF-α) production generated by the lipopolysaccharide (LPS). All compounds revealed slight cytotoxic properties with the inhibition of PBMC. The most promising derivatives 20–22 (22 containing the acetamidopropyl substituent) were screened against the leukemia L-1220 and the colon carcinoma SW-948 cell lines. The most effective compound turned out the parent compound 20, exhibiting similar effect as cisplatin against the cancer cells of carcinoma SW-948 and leukemia L-1210 at 5 and 10 µg/mL, respectively.
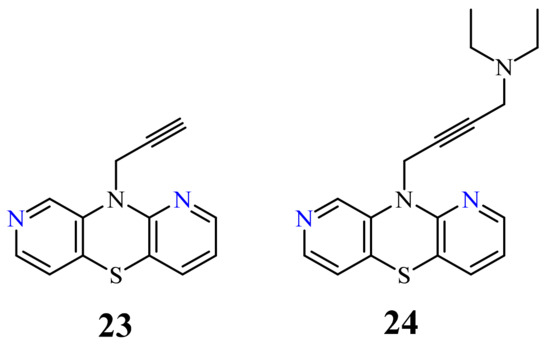
Another group of 1,8-diazaphenothiazines was obtained by a transformation of the 10-propargyl-1,8-diazaphenothiazine (23) (Figure 11) into the dialkylaminobutynyl derivatives. Its anticancer activity was evaluated against the cell lines of human glioblastoma SNB-19, melanoma C-32, and breast cancer T-47D using cisplatin as a reference compound. The compounds were characterized by a moderate activity (IC50 in the range of 26 to 46 µg/mL) which was dependent on both the type of substituent and the type of cell line. The most active compound was 10-diethylaminobutynyl-1,8-diazaphenothiazine (24) (IC50 = 26.1 µg/mL) against the melanoma C-32 [45].
In order to search for quantitative relationships between physicochemical and pharmacological properties of the investigated compounds, preliminary QSAR (Quantitative Structure-Activity Relationship) studies were undertaken for anticancer and immunosuppressant 1,8-diazaphenothiazines. For this group of diazaphenothiazines, some correlations between the TNF-α inhibition, anticancer activity, and lipophilicity were observed [78].
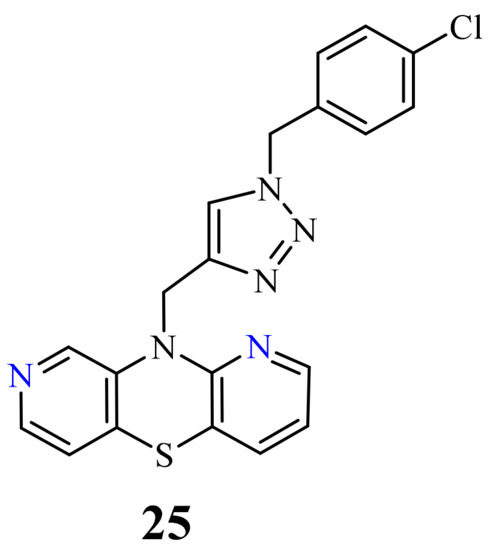
A group of novel hybrids of the 1,8-diazaphenothiazine and 1,2,3-triazole systems were obtained and tested for their cytotoxicity against the glioblastoma SNB-19, colorectal carcinoma Caco-2, lung cancer A549, and breast cancer MDA-MB231 cell lines. The anticancer activity of this group of derivatives was differential (IC50 = 1.8–49 µM). The compound 25 (Figure 12) with the p-chlorobenzyl substituent showed a strong promising anticancer activity (IC50 = 1.82 µM) in relation to the lung cancer A549. However, this activity was lower than that of the isomeric 1,6-diazaphenothiazine 19 (IC50 = 0.25 µM) [69].
3.3. 1,9-Diazaphenothiazines (Dipyrido[2,3-b;2’,3’-e][1,4]thiazines).
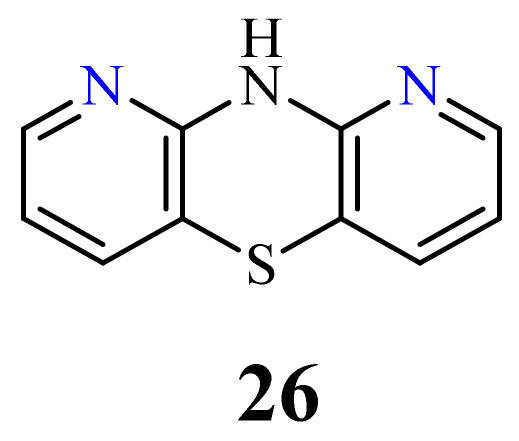
The structure of 10H-1,9-diazaphenothiazine (26) (Figure 13) was first described in a US patent in 1957. However, the information contained in this report was very cursory and did not present the biological aspects of this compound [79].
Very recently, an efficient synthesis of 10H-1,9-diazaphenothiazine (26) was described, along with a complete structural analysis, as well as new 10-substituted derivatives containing alkyl, alkynyl, and dialkylaminoalkyl substituents. These compounds with various substituents were screened for their anticancer activity against using the glioblastoma SNB-19, melanoma C-32, and breast cancer MDA-MB-231 cell lines [72]. The parent compound 10H-1,9-diazaphenothiazine (26) was very active against melanoma C-32 (IC50 = 3.83 µM), even more potent than cisplatin (IC50 = 13.2 µM), but inactive against other lines. These derivatives induced a varying anticancer activity. The most promising derivatives in this group were compounds 27 and 28 with the propynyl and diethylaminoethyl groups (Figure 14).

10-Propynyl-1,9-diazaphenothiazine (27) was highly active against the glioblastoma SNB-19 (IC50 = 3.85 µM) and the melanoma C-32 (IC50 = 3.37 µM). 10-Diethylaminoethyl-1,9-diazaphenothiazine (28) was even more active against the glioblastoma SNB-19 with an IC50 value of 0.34 µM, and the breast cancer MDA-MB-231 cell line with IC50 = 2.13 µM. For those two compounds, the expression of H3, TP53, CDKN1A, BCL-2, and BAX genes was detected by the RT-qPCR method. The analysis of the gene expression revealed that both compounds selectively reduced the expression of H3 and TP53, and enhanced the expression of CDKN1A in the examined cell lines. The gene expression ratio of BAX/BCL-2 indicated the induction of mitochondrial apoptosis in two cancer cell lines (SNB-19 and MDA-MB-231). In the melanoma C-32 cell line, the transcriptional gene activity suggests a different way of cell death. The proteome profiling study showed the most probable compound action on SNB-19 cells through the intrinsic mitochondrial pathway of apoptosis [72].
3.4. 2,7-Diazaphenothiazines (Dipyrido[3,4-b;3’,4’-e][1,4]thiazines)
A family of 2,7-diazaphenothiazines was widely described in the literature both due to good chemical properties and highly significant bioactive properties. The first reports on the synthesis of this new heterocyclic system appeared in the literature at the beginning of the 21st century [80][81].
The parent compound 10H-2,7-diazaphenothiazine (29) and its methyl derivative 30 (Figure 15) strongly reduced the proliferative response of PBMC to PHA at 10 µg/mL. The former derivative exhibited very suppressive effect (72% at 1 µg/mL) in relation to the secondary humoral response to sheep red blood cells (SRBC). In addition, this derivative importantly reduced the delayed-type hypersensitivity (DTH) response to ovalbumin in the in vivo test using mice. In the experiment of cytokine production induced by LPS, this compound totally inhibited the IL-6 production at 100 µg/mL and moderately inhibited the TNF-α production at 10 µg/mL. The group of tested 2,7-diazaphenothiazines had low toxicity toward mouse splenocytes [82].
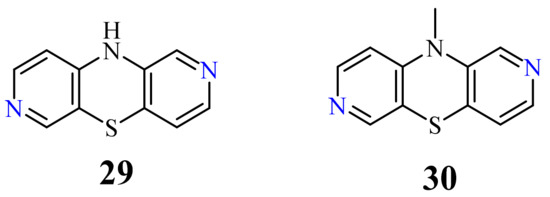
10H-2,7-diazaphenothiazine (29), and its selected 10-substituted derivatives containing various substituents, were also screened for their anticancer activity in the National Cancer Institute, Bethesda, MD, USA. In a preliminary experiment, the most promising compound turned out to be parent compound 29, which was screened on about 60 cell lines including leukemia, melanoma, non-small cell cancer, colon cancer, CNS cancer, ovarian cancer, renal cancer, prostate cancer, and breast cancer. The strongest action was observed with relation to the non-small lung cancer cell lines HOP-62 and HOP-92 with IC50 values of 0.3 and 1.7 µg/mL, respectively. Other types of cancer cell lines were also sensitive to compound 29 showing the following IC50 values: 2.4 and 3.6 µg/mL (colon 205 and HCT-116), 3.1, 3.9, and 5.4 µg/mL (renal RXF 393, 736-0 and ACHN); 4.1 µg/mL (leukemia HL-60(TB)); 5.9 µg/mL (breast HS 578T), 6.5 µg/mL (melanoma M-14); 6.8 µg/mL (CNS SF-539 and SNB-19); 7.1 µg/mL (ovarian OVCAR-8); and 8.4 µg/mL (prostate PC-3). 10-Substituted derivatives were less active in the preliminary test [73][83].
10H-2,7-diazaphenothiazine (29) and its nitrophenyl and pyrrolidynyloethyl derivatives exhibited a meaningful antioxidant activity (IC50 = 64–125 µM) in the experiment of non-enzymatic peroxidation of hepatic microsomal membrane lipids [84].
Another group of 2,7-diazaphenothiazine derivatives was obtained by a transformation of the 10-propargyl derivative of 2,7-diazaphenothiazine into the dialkylaminobutynyl derivatives. The anticancer activity was evaluated against the cell lines of human glioblastoma SNB-19, melanoma C-32, and breast cancer T-47D. The most active compound was the derivative 31 with the methylpiperazinylbutynyl substituent (Figure 16) against the carcinoma T-47D with the IC50 values of 9.6 µg/mL, being more potent than cisplatin (46.9 µg/mL). This derivative was also the most effective in relation to the glioblastoma SNB-19 (21.2 µg/mL). The remaining derivatives of this group showed a moderate activity with IC50 values in the range of 24.8 to 29.2 µg/mL. To understand the anticancer mechanism, the effect of derivative 31 on the expression of genes encoding TP53, CDKN1A, BCL-2, and BAX in the tested cancer cells was investigated. The RT-qPCR experiment showed the heightened number of CDKN1A copies in the T-47D and SNB-19 cells which implied the ability of the cell cycle arrest. Examination of the ratio of gene expression BCL-2/BAX revealed the activation of mitochondrial apoptosis in the T-47D cells [45].
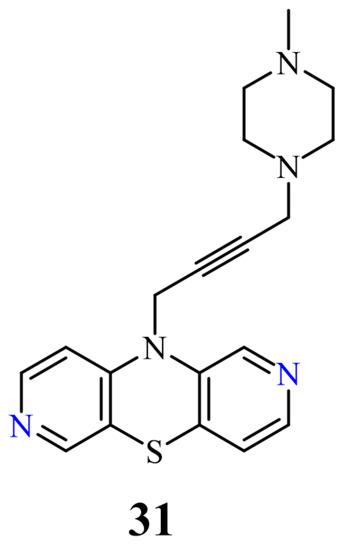
The hybrids of 2,7-diazaphenothiazine and 1,2,3-triazole systems were obtained and examined for their cytotoxicity against the glioblastoma SNB-19, colorectal carcinoma Caco-2, lung cancer A549, and breast cancer MDA-MB231 cell lines (Figure 17). The anticancer activity of this group of derivatives was in the range IC50 = 0.26–49 μΜ. The most active compound was the derivative with benzyl substituent 32 in the 1,2,3-triazole ring showing the IC50 values of 0.26 μΜ against the Caco-2 and A549 cancer cell lines and 0.77 μΜ against the MDA-MB231 cancer cell line. A similarly high antiproliferative activity exhibited the derivative 33 with p-fluorobenzyl substituent 33 in 1,2,3-triazole ring against the MDA-MB231 (IC50 = 0.64 μΜ) and against the A549 (IC50 = 0.65 μΜ) cancer cell line [69].
3.5. 3,6-Diazaphenothiazines (Dipyrido[2,3-b;4’,3’-e][1,4]thiazines)
The 3,6-diazaphenothiazine system with the nitro group in position 1 and the chlorine atom and methoxy group in position 7 was described in the 1960s, but the identification of these compounds was based only on the logic of the conducted chemical syntheses. The author drew attention to the chemical properties of these molecules, while the biological activities were not described [86][87].
In 2016, a novel group of 3,6-diazaphenothiazines was described both due to interesting chemical and structural properties, but above all anticancer properties [46]. 10H-3,6-Diazaphenothiazine (34) (Figure 18) and its 10-derivatives containing varied substituents were tested for their anticancer activity. The parent compound 34 exhibited an extremely strong action against the glioblastoma SNB-19, melanoma C-32, and breast cancer MCF-7 cell lines with the IC50 values of 0.46 and 0.72 µg/mL. The derivative 35 (containing the 2-pyrimidinyl substituent, see Figure 18) showed a powerful activity against the breast cancer MCF-7 with the IC50 value of 0.73 µg/mL. Both derivatives were more effective than cisplatin. The derivative 36 (with the dimethylaminopropyl substituent) exhibited similar action to cisplatin (6.3 vs. 7.8 µg/mL) against the melanoma C-32 and moderate activity (11.3 µg/mL) against the breast cancer MCF-7. The family of these diazaphenothiazines showed non-toxic or almost non-toxic action against the normal fibroblast HFF-1 cell line [46].
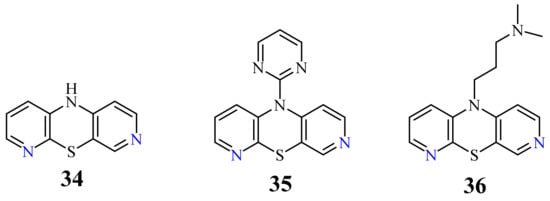
Anticancer activity of 10H-3,6-diazaphenothiazine (34) was also studied on the A2780 ovarian cancer cells by an investigation on cytotoxicity profiles, the mechanism of apoptosis, and cell invasion. This parent compound induced a dose-dependent inhibition on the A2780 cancer cells (IC50 = 0.62 μM), with a significant less cytotoxicity towards the normal kidney HEK293 cells and the normal heart H9C2 cells. It was presented that the generation of reactive oxygen species (ROS) and the polarization of mitochondrial membrane potential (ΔΨm) suggested that compound 34 induced cell death through the oxidative damage. This compound elicited an upregulation of caspase-6, -3, and -7, which are actively involved in the formation of cell shrinkage, chromatin condensation, and the fragmentation of DNA. Additionally, the activation of caspase-3 brought about an increased enzymatic activity of DFFA (DNA fragmentation factor-α). This compound induced apoptosis via an intrinsic (mitochondria-dependent) and an extrinsic (cell death receptor-dependent) pathway. The inhibition of NF-κB and the subsequent inhibition of BIRC6-XIAP complexes reduced the invasion rate of the A2780 cancer cells penetrating through the MatrigelTM Invasion Chamber. The investigated compound exhibited a cytostatic action and significantly arrested cell proliferation at the G2/M phase. The presented results suggested that the 10H-3,6-diazaphenothiazines (34) would become promising chemotherapeutic agents in the future [74].
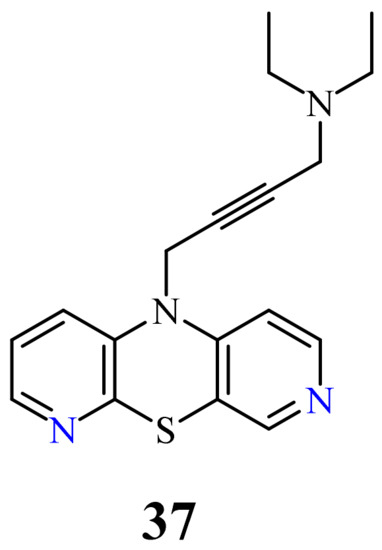
Very recently, another group of new 10-substituted derivatives of 3,6-diazaphenothiazine, containing the triple bond linker ended with tertiary cyclic and acyclic amine groups was synthesized. This group exhibited varied anticancer activities against the human glioblastoma SNB-19, melanoma C-32, and breast cancer MDA-MB231 cancer lines, depending on the nature of the substituents. The most active compound was the derivative 37 with the diethylamino-2-butynyl substituent (Figure 19) against the glioblastoma SNB-19 (IC50 = 0.11 μg/mL). The analysis of genes expressions (H3, TP53, CDKN1A, BCL-2, and BAX) using the RT-qPCR method indicated the induction of mitochondrial apoptosis in the SNB-19 cells [75].
At the same time, novel 1,2,3-triazole-dipyridothiazine hybrids containing the 3,6-diazaphenothiazine system and selected p-substituted benzyl substituents at the 1,2,3-triazole ring were synthesized and screened for the anticancer activity. In this group of derivatives, the compound 38 with the p-fluorobenzyl substituent showed a very promising anticancer action (IC50 = 0.25 µM) in relation to the colorectal carcinoma Caco-2 and the lung cancer A549 (Figure 20). Additionally, the compound was nontoxic against the normal fibroblasts NHDF in the range of tested concentration [69].
In the group of 3,6-diazaphenothiazines, extensive studies of lipophilicity parameters and ADME properties were performed in order to look for the lipophilicity–activity relationship [83]. Nonetheless, a simple answer was not obtained as to which factor directly determines the high antitumor activity. The conducted research did not show a direct dependence of the anticancer activity on the lipophilic properties of this group of dipyridothiazines.
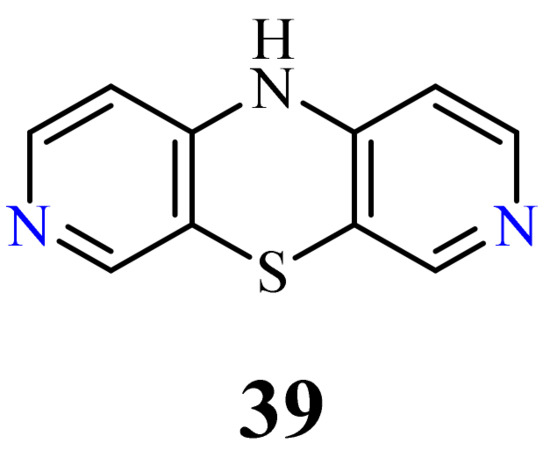
3.6. 3,7-Diazaphenothiazines (Dipyrido[3,4-b;4’,3’-e][1,4]thiazines)
This group of diazaphenothiazines was described in the 1960s [88][89][90][91]. The parent compound 10H-3,7-diazaphenothiazine (39) and its 10-derivatives containing the diethylaminoethyl and dimethylaminopropyl substituents (Figure 21) exerted an antihistaminic action. Unfortunately, these activities were not substantially better than those of the leading phenothiazine drugs in the pharmaceutical market at the time. The anticancer activity of these compounds has not yet been evaluated.
Out of the six types of isomeric dipyridothiazines, five types were tested successfully against various kinds of cancer cell lines.
Most of the biological results discussed in this chapter were obtained by the authors in cooperation with international and domestic research groups.
The information provided on anticancer activity of pyridobenzothiazines and dipyridothiazines was summarized in Table 1.
References
- Reinhardt, C.; Travis, A.S. Heinrich Caro and the Creation of Modem Chemical Industry, Chemists and Chemistry; Springer Science + Business Media: Dordrecht, The Netherlands, 1998; Volume 19, pp. 242–245.
- Bernthsen, A. Ű̈ber das Methylenblau. Ber. Dtsch. Chem. Ges. 1883, 16, 2896–2904.
- Gupta, R.R.; Kumar, M. Synthesis, properties and reactions of phenothiazines. In Phenothiazine and 1,4-Benzothiazines—Chemical and Biological Aspect; Gupta, R.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1988; pp. 1–161.
- Ohlow, M.J.; Moosmann, B. Foundation review: Phenothiazine: The seven lives of pharmacology’s first lead structure. Drug Discov. Today 2011, 16, 119–131.
- Huang, J.; Zhao, D.; Liu, Z.; Liu, F. Repurposing psychiatric drugs as anti-cancer agents. Cancer Lett. 2018, 419, 257–265.
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.E.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198.
- Wu, C.-H.; Bai, L.-Y.; Tsai, M.-H.; Chu, P.-C.; Chiu, C.-F.; Chen, M.Y.; Chiu, S.-J.; Chiang, J.-H.; Weng, J.-R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents—A drug repurposing strategy. Sci. Rep. 2016, 6, 1–16.
- Kristiansen, J.E.; Dastidar, S.G.; Palchoudhuri, S.; Roy, D.S.; Das, S.; Hendricks, O.; Christensen, J.B. Phenothiazines as a solution for multidrug resistant tuberculosis: From the origin to present. Int. Microb. 2015, 18, 1–12.
- Amaral, L.; Viveiros, M. Thioridazine: A non-antibiotic drug highly effective, in combination with first line anti-tuberculosis drugs, against any form of antibiotic resistance of mycobacterium tuberculosis due to its multi-mechanisms of action. Antibiotics 2017, 6, 1.
- Sharma, S.; Singh, A. Phenothiazines as anti-tubercular agents: Mechanistic insights and clinical implications. Expert Opin. Investig. Drugs 2011, 20, 1665–1676.
- Sellamuthu, S.; Bhat, M.F.; Kumar, A.; Singh, S.K. Phenothiazine: A better scaffold against tuberculosis. Mini-Rev. Med. Chem. 2017, 17, 1442–1451.
- Spengler, G.; Csonka1, Á.; Molnár, J.; Amaral, L. The anticancer activity of the old neuroleptic phenothiazine-type drug thioridazine. Anticancer Res. 2016, 36, 5701–5706.
- Varga, B.; Csonka, Á.; Csonka, A.; Molnár, J.; Amaral, L.; Spengler, G. Possible biological and clinical applications of phenothiazines. Anticancer Res. 2017, 37, 5983–5993.
- Sudeshna, G.; Parimal, K. Muliple non-psychiatric effect of phenothiazines: A review. Eur. J. Pharmacol. 2010, 648, 6–14.
- Yue, H.; Huang, D.; Qin, L.; Zheng, Z.; Hua, L.; Wang, G.; Huang, J.; Huang, H. Targeting lung cancer stem cells with antipsychological drug thioridazine. BioMed Res. Int. 2016.
- Nagy, S.; Argyelan, G.; Molnár, J.; Kawase, M.; Motohashi, N. Antitumor activity of phenothiazine-related compounds. Anticancer Res. 1996, 16, 1915–1918.
- Mosnaim, A.D.; Ranade, V.V.; Wolf, M.E.; Puente, J.; Valenzuela, M.A. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am. J. Therapeut. 2006, 13, 261–273.
- Pluta, K.; Jeleń, M.; Morak-Młodawska, B.; Zimecki, M.; Artym, J.; Kocięba, M.; Zaczyńska, E. Azaphenothiazines a promising phenothiazine derivatives. An insight into nomenclature, synthesis, structure elucidation and biological properties. Eur. J. Med. Chem. 2017, 138, 774–806.
- Pluta, K.; Morak-Młodawska, B.; Jeleń, M. Synthesis and properties of diaza-, triaza- and tetraazaphenothiazines. J. Heterocycl. Chem. 2009, 46, 355–391.
- Motohashi, N. Antitumor activity of phenothiazines (phenothiazine oncology). In Phenothiazines and 1,4-Benzothiazines. Chemical and Biological Aspects; Gupta, R.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1988; pp. 705–770.
- Sakagami, H.; Takahashi, H.; Yoshida, H.; Yamamura, M.; Fukuchi, K.; Gomi, K.; Motohashi, N.; Takeda, M. Induction of DNA fragmentation in human Myelogenous Leukaemic cell lines by phenothiazine-related compounds. Anticancer Res. 1995, 15, 2533–2540.
- Motohashi, N.; Sakagami, H.; Kamata, K.; Yamamoto, Y. Cytotoxicity and differentiation-inducing activity of phenothiazine and benzo[a]phenothiazine derivatives. Anticancer Res. 1991, 11, 1933–1937.
- Wuonola, M.A.; Palfreyman, M.G.; Motohashi, N.; Kawase, M.; Gabay, S.; Gupta, R.R.; Molnár, J. The primary in vitro anticancer activity of “half-mustard type” phenothiazines in NCI’s revised anticancer screening paradigm. Anticancer Res. 1998, 18, 337–348.
- Motohashi, N.; Kurihara, T.; Sakagami, H.; Szabo, D.; Csuri, K.; Molnár, J. Chemical structure and tumor type specificity of “half-mustard type” phenothiazines. Anticancer Res. 1999, 19, 1859–1864.
- Motohashi, N.; Kawase, M.; Kurihara, T. Synthesis and antitumor activity of 1-[2(chloroethyl)-3-(-substituted-10H-phenothiazin-10-yl)]alkyl-1-ureas as potent anticancer agents. Anticancer Res. 1996, 16, 2525–2532.
- Motohashi, N.; Kawase, M.; Saito, S. Synthesis and biological activity of N-acylphenothiazines. Int. J. Antimicrob. Agents. 2000, 14, 203–207.
- Motohashi, N.; Kawase, M.; Saito, S.; Sakagami, H. Antitumor potential and possible targets of phenothiazine-related compounds. Curr. Drug Targets 2000, 1, 237–245.
- Motohashi, N.; Kawase, M.; Satoh, K.; Sakagami, H. Cytotoxic potential of phenothiazines. Curr. Drug Targets 2006, 7, 1055–1066.
- Gaye-Seye, M.D.; Aaron, J.J.; Parkanyi, C.; Motohashi, N. Luminescence and photophysical properties of benzo[a]phenothiazines-therapeutic, physico-chemical and analytical applications. Curr. Drug Targets 2006, 7, 1083–1093.
- Bisi, A.; Meli, M.; Gobbi, S.; Rampa, A.; Tolomeo, M.; Dusonchet, L. Multidrug resistance reverting activity and antitumor profile of new phenothiazine derivatives. Bioorg. Med. Chem. 2008, 16, 6474–6482.
- Khandelwal, N.; Yadav, A.; Gautam, N.; Gautam, D.C. Study and synthesis of biologically active phenothiazines, their sulfones, and ribofuranosides. Nucleos. Nucleot. Nucl. Acids 2012, 31, 680–691.
- Gautam, N.; Garg, A.; Lal, T.; Gautam, D.C.; Joshi, J. Synthesis and antimicrobial assessment of new substituted 10H-phenothiazines, their sulfone derivatives, and ribofuranosides. Heterocycl. Commun. 2014, 20, 343–349.
- Prinz, H.; Ridder, A.-K.; Vogel, K.; Böhm, K.J.; Ivanov, I.; Ghasemi, J.B.; Aghaee, E.; Müller, K. N-Heterocyclic (4-phenylpiperazin-1-yl)methanones derived from phenoxazine and phenothiazine as highly potent inhibitors of tubulin polymerization. J. Med. Chem. 2017, 60, 749–766.
- Zhang, J.-X.; Guo, J.-M.; Zhang, T.-T.; Lin, H.-J.; Qi, N.-S.; Li, Z.-G.; Zhou, J.-C.; Zhang, Z.-Z. Antiproliferative phenothiazine hybrids as novel apoptosis inducers against MCF-7 breast cancer. Molecules 2018, 23, 1288.
- Krishnan, K.G.; Kumar, C.U.; Lim, W.-M.; Mai, C.-W.; Thanikachalam, P.V.; Ramalingan, C. Novel cyanoacetamide integrated phenothiazines: Synthesis, characterization, computational studies and in vitro antioxidant and anticancer evaluations. J. Mol. Struct. 2020, 1199, 127037.
- Pluta, K.; Morak-Młodawska, B.; Jeleń, M. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 2011, 46, 3179–3189.
- Gopi, C.; Dhanaraju, M.D. Recent progress in synthesis, structure and biological activities of phenothiazine derivatives. Rev. J. Chem. 2019, 9, 95–126.
- Gao, Y.; Sun, T.-Y.; Bai, W.-F.; Bai, C.-G. Design, synthesis and evaluation of novel phenothiazine derivatives as inhibitors of breast cancer stem cells. Eur. J. Med. Chem. 2019, 183, 111692.
- Yale, H.L.; Bernstein, J. Azaphenothiazine Compound and Their Preparation. U.S. Patent 2943086, 6 May 1960.
- Uhrig, S.; Hirth, N.; Broccoli, L.; Von Wilmsdorff, M.; Bauer, M.; Sommer, C.; Zink, M.; Steiner, J.; Frodl, T.; Malchow, B.; et al. Reduced oxytocin receptor gene expression and binding sites in different brain regions in schizophrenia: A post-mortem study. Schizophr. Res. 2016, 177, 59–66.
- Winkler, D.; Pjrek, E.; Lanzenberger, R.; Baldinger, P.; Eitel, D.; Kasper, S.; Frey, R. Actigraphy in patients with treatment-resistant depression undergoing electroconvulsive therapy. J. Psychiatr. Res. 2014, 57, 96–100.
- Kleinmann, A.; Schrader, V.; Stübner, S.; Greil, W.; Kahl, K.G.; Bleich, S.; Grohmann, R.; Frieling, H.; Toto, S. Psychopharmacological treatment of 1650 in-patients with acute mania-data from the AMSP study. J. Affect. Disord. 2016, 191, 164–171.
- Scharfetter, J.; Fischer, P. QTc prolongation induced by intravenous sedation with Haloperidol, Prothipendyl and Lorazepam. Neuropsychiatrie 2014, 28, 1–5.
- Declercq, T.; Petrovic, M.; Azermani, M.; Vander Stichle, R.; De Sutter, A.I.; Van Driel, M.L.; Christiaens, T. Withdrawal versous continuation of chronic antipsychotic drugs for behavioral and psycholofical symptoms in older people with dementia. Cochrane Database Syst. Rev. 2013, 28, 1–95.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M.; Kuśmierz, D. Synthesis and anticancer and lipophilic properties of 10-dialkylaminobutynyl derivatives of 1,8- and 2,7-diazaphenothiazines. J. Enzym. Inhib. Med. Chem. 2016, 31, 1132–1138.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Suwińska, K.; Jeleń, M.; Kuśmierz, D. 3,6-Diazaphenothiazines as potential lead molecules—Synthesis, characterization and anticancer activity. J. Enzym. Inhib. Med. Chem. 2016, 31, 1512–1519.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M. Synthesis, spectroscopic characterization, and anticancer activity of new 10-substituted 1,6-diazaphenothiazines. Med. Chem. Res. 2016, 25, 2425–2433.
- Kushwaha, K.; Kaushik, N.K.; Kaushik, N.; Chand, M.; Kaushik, R.; Ha Choi, E.; Jain, S.C. Novel aminoalkylated azaphenothiazines as potential inhibitors of T98G, H460 and SNU80 cancer cell lines in vitro. Bioorg. Med. Chem. Lett. 2016, 26, 2237–2244.
- Kaur, P.; Hann Chu, J.J. Chikungunya virus: An update on antiviral development and challenges. Drug Discov. Today 2013, 18, 969–983.
- Hagerdorn, H.W.; Zuck, S.; Schulz, R. Prothipendyl: Detection and elimination in the horse—A casereport. Dtsch Tierarztl Wochenschr 1996, 103, 125–127.
- Martindale, the Extra Pharmacopoeia, 29th ed.; Reynolds, J.E.F. (Ed.) Pharmaceutical Press: London, UK, 1989.
- Shaikh, S.M.T.; Seetharamappa, J.; Kandagal, P.B.; Ashoka, S. Binding of the bioactive component isothipendyl hydrochloride with bovine serum albumin. J. Mol. Struct. 2006, 786, 46–52.
- Moreau, A.; Dompmartin, A.; Dubreuil, A.; Leroy, D. Phototoxic and photoprotective effects of topical isothipendyl. Photodermatol. Photoimmunol. Photomed. 1995, 11, 50–54.
- Bibas, N.; Sartor, V.; Bulai Livideanu, C.; Bagheri, H.; Nougue, J.; Giordano- Labadie, F.; Maza, A.; Paul, C.; Chouini-Lalanne, N.; Marguery, M.C. Contact photoallergy to isothipendyl chlorohydrate. Dermatology 2012, 224, 289–291.
- Amin, A.S.; El-Sheikh, R.; Zahran, F.; El-fetough Gouda, A.A. Spectrophotometric determination of pipazethate HCl, dextromethorphan HBr and drotaverine HCl in their pharmaceutical preparations. Spectrochim. Acta A 2007, 67, 1088–1093.
- Atkinson, E.R.; Russ, P.L.; Tucker, M.T. Neuropharmacological profile of 1-azaphenothiazine-10-thiolcarboxylates. J. Med. Chem. 1971, 14, 1005–1007.
- Sharma, A.; Tyagi, E. Synthesis of some substituted pyrido[3,2-b][1,4]benzothiazines and their antibacterial activity. Pharmazie 1991, 46, 746–747.
- Swati, S.S.; Mishira, A.K.; Prakash, L. Synthesis of some novel 1-azaphenothiazines and their mesoionic as analogues of popent CNS-depressants. Phosphorus Sulfur Silicon Relat. Elem. 1996, 117, 111–120.
- Agrawal, H.; Yador, A.K.; Prakash, L. An elegant synthesis of some new potential biologically active pyrido[3,3-b][1,4]benzothiazine derivatives and their nucleosides by phase transfer catalysis. Heterocycl. Commun. 1998, 4, 589–594.
- Kumar, N.; Singh, G.; Khatoon, S.; Yadav, A. Synthesis and antimicrobial activities of novel 10H-pyrido[3,2-b][1,4]benzo[b]thiazine ribofuranosides. Indian J. Chem. B 2003, 42, 2015–2018.
- Madrid, P.; Polgar, W.; Toll, L.; Tanga, M. Synthesis and antitubercular activity of phenothiazines with reduced binding to dopamine and serotonin receptors. Bioorg. Med. Chem. Lett. 2007, 17, 3014–3017.
- Saggiomo, A.; Craig, P.; Gordon, M. Synthesis of 2-aza- and 8-chloro-2-aza-phenothiazine. J. Org. Chem. 1958, 23, 1906–1909.
- Okafor, C.O. Studies in the heterocyclic series. III. The Chemistry of azaphenothiazine compounds. Int. J. Sulfur Chem. B 1971, 6, 239–265.
- Zirkle, C.L.; Kaiser, C. Antipsychotic agents (tricyclic), In Psychopharmacological Agents; Gordon, M., Ed.; Academic Press: New York, NY, USA, 1974; Volume 3, pp. 39–128.
- Clarke, F.H.; Silverman, G.B.; Watnick, C.M.; Sperber, N. 3-Azaphenothiazine and dialkylaminoalkyl derivatives. J. Org. Chem. 1961, 26, 1126–1132.
- Chorvat, R.J.; Desai, B.N.; Radak, S.E.; Bloss, J.; Hirsch, J.; Tenen, S. Synthesis, benzodiazepine receptor binding, and anticonvulsant activity of 2,3-dihydro-3-oxo-5H-pyrido[3,4-b][1,4]benzothiazine-4-carbonitriles. J. Med. Chem. 1983, 26, 845–850.
- Rhone-Poulenc, Azaphenothiazines and Intermediates. British Patent 791190, 26 February 1958.
- Yamada, K.; Miyamoto, M.; Tatsuya, T.; Sato, K.; Soejima, M.; Sato, T.; Kikuchi, K.; Yoshimura, H.; Moriya, K.; Sakuma, Y. Preparation of Heterocycle-Fused Benzothiazine Derivatives as Allergy Inhibitors. Japan Patent WO 9943683 A1 19990909, 2 September 1999.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M.; Kuśmierz, D. Design, Synthesis, and Structural Characterization of Novel Diazaphenothiazines with 1,2,3-Triazole Substituents as Promising Antiproliferative Agents. Molecules 2019, 24, 4388.
- Morak-Młodawska, B.; Pluta, K.; Zimecki, M.; Jeleń, M.; Artym, J.; Kocięba, M. Synthesis and selected immunological properties of 10-substituted 1, 8-diazaphenothiazines. Med. Chem. Res. 2015, 24.
- Pluta, K.; Morak-Młodawska, B.; Zimecki, M.; Jeleń, M.; Artym, J.; Kocięba, M. 10H-1,8-Diazaphenothiazine, Its 10-Substituted Derivatives, Their Usage, the Way of Synthesis and Their Pharmaceutical Compositions. Polish Patent PL 227918 B1, 10 July 2013.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M.; Kuśmierz, D.; Suwińska, K.; Shkurenko, A.; Czuba, Z.; Jurzak, M. 10H-1,9-diazaphenothiazine and its 10-derivatives: Synthesis, characterisation and biological evaluation as potential anticancer agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 1298–1306.
- Pluta, K.; Jeleń, M.; Morak-Młodawska, B.; Zimecki, M.; Artym, J.; Kocięba, M. Anticancer activity of newly synthesized azaphenothiazines in NCI’s anticancer screening. Pharmacol. Rep. 2010, 62, 319–332.
- Zhang, J.; Chen, M.; Wenzhi, Z.; Okechukwu, P.N.; Morak-Młodawska, B.; Pluta, K.; Jeleń, M.; Md Akim, A.; Ang, K.-P.; Ooi, K.K. 10H-3,6-Diazaphenothiazines Induce G2/M Phase Cell Cycle Arrest, Caspase-dependent Apoptosis and Inhibits Cell Invasion of A2780 Ovarian Carcinoma Cells through Regulation on NF-κB and [BIRC6-XIAP] Complexes. Drug Des. Dev. Ther. 2017, 11, 3045–3063.
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M.; Kuśmierz, D. Synthesis, anticancer activity and apoptosis induction of novel 3,6-diazaphenothiazines. Molecules 2019, 24, 267.
- Rodig, O.; Collier, R.; Schlatzer, R. Pyridine chemistry. II. Further studies on the Smiles rearrangement of the 3-amino-2,2‘-dipyridyl sulfide system. The synthesis of some 1,6-diazaphenothiazines. J. Med. Chem. 1965, 9, 116–120.
- Morak-Młodawska, B.; Pluta, K.; Jeleń, M. Lipophilicity of New Anticancer 1,6- and 3,6-diazaphenothiazines by of Use RP TLC and Different Computational Methods. J. Chrom. Sci. 2018, 1–6.
- Morak-Młodawska, B.; Pluta, K.; Jeleń, M. Estimation of the Lipophilicity of New Anticancer and Immunosuppressive 1,8-Diazaphenothiazine Derivatives. J. Chrom. Sci. 2015, 53, 462–466.
- Rath, S. Dimethylaminopropyldipyridothiazine. U.S. Patent 2,789,978, 23 April 1957.
- Morak, B.; Pluta, K.; Suwińska, K. Unexpected simple route to novel dipyrido-1,4-thiazine system. Heterocyclic Commun. 2002, 8, 331–334.
- Morak, B.; Pluta, K. Synthesis of novel dipyrido-1,4-thiazines. Heterocycles 2007, 71, 1347–1361.
- Zimecki, M.; Artym, J.; Kocięba, M.; Pluta, K.; Morak-Młodawska, B.; Jeleń, M. The immunosupressive activities of newly synthesized azaphenothiazines in human and mouse models. Cell. Mol. Biol. Lett. 2009, 14, 622–635.
- Pluta, K.; Jeleń, M.; Morak-Młodawska, B. Anticancer activity of selected dipyridothiazines and diquinothiazines determined in National Cancer Institute, in Bethesdzie, USA. Farm. Przegląd Nauk. 2009, 10, 26–29.
- Morak-Młodawska, B.; Pluta, K.; Matralis, A.N.; Kourounakis, A.P. Antioxidant activity of newly synthesized 2,7-diazaphenothiazines. Archiv. Pharm. Chem. Life Sci. 2010, 343, 268–273.
- Morak-Młodawska, B.; Pluta, K.; Jeleń, M. Evaluation of the Lipophilicity of New Anticancer 1,2,3-Triazole-Dipyridothiazine Hybrids Using RP TLC and Different Computational Methods. Processes 2020, 8, 858.
- Okafor, C. Studies in heterocyclic series. I. A novel diazaphenothiazine system. J. Org. Chem. 1967, 32, 2006–2007.
- Okafor, C. Heterocyclic series. II. 3,6-Diazaphenothiazine sulfoxides and other potential antiparasitic and pesticidal agents. J. Chem. Eng. Data 1971, 16, 244–246.
- Kopp, E.; Strell, M. Űber 2,7-diazaphenothiazin. Reaktionen in der Pyridinreihe, IV. Arch. Pharm. 1962, 295, 99–106.
- Strell, M.; Kopp, E.; Janson, R. Verfahren zur Herstellung von 2,7-Diazaphenothiazinen. German Patent DE 1 147 235, 18 April 1963.
- Kopp, E. Tautomerie an einem 2, 7‐Diazaphenothiazinderivat. Reaktionen in der Pyridinreihe, V. Arch. Pharm. 1962, 295, 561–564.
- Werle, E.; Kopp, E.; Leysath, G. Arzneim-Forsch Die Antihistaminwirkung von 2,7-Diazaphenothiazin und einiger seiner derivate. Arzneim-Forsch 1962, 4, 443–444.


