 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucy Baldeón | + 3506 word(s) | 3506 | 2021-02-20 10:36:31 | | | |
| 2 | Lily Guo | Meta information modification | 3506 | 2021-03-08 04:36:06 | | |
Video Upload Options
In December 2019, a novel coronavirus known as SARS-CoV-2 was first detected in Wuhan, China, causing outbreaks of the coronavirus disease COVID-19 that has now spread globally. For this reason, The World Health Organization (WHO) declared COVID-19 a public health emergency in March 2020. People living with pre-existing conditions such as obesity, cardiovascular diseases, type 2 diabetes (T2D), and chronic kidney and lung diseases, are prone to develop severe forms of disease with fatal outcomes. Metabolic diseases such as obesity and T2D alter the balance of innate and adaptive responses. Both diseases share common features characterized by augmented adiposity associated with a chronic systemic low-grade inflammation, senescence, immunoglobulin glycation, and abnormalities in the number and function of adaptive immune cells. In obese and T2D patients infected by SARS-CoV-2, where immune cells are already hampered, this response appears to be stronger.
1. Introduction
Outbreak of the Coronavirus disease in 2019 (COVID-19) has spread rapidly across the world evidencing the weakness of the public health system worldwide [1][2]. Free access to scientific information related to COVID-19, facilitates the understanding of epidemiological, clinical, and molecular aspects of the new virus, which can help to contain the disease and to give alternatives for treatment [3]. A beta-coronavirus SARS-CoV-2 was identified as the causative of COVID-19. On 11 March due to high spread, infectious potential, morbidity, and mortality, the World Health Organization (WHO) declared COVID-19 as a global pandemic [4]. As of January 2021, over 85 million cases and nearly 2 million deaths worldwide have been reported [5].
The majority of infected patients experience mild to moderate symptoms such as fever, headache, cough, myalgia, and diarrhea [6][7]. However, people living with pre-existing conditions such as obesity, cardiovascular diseases, type 2 diabetes (T2D), chronic kidney and lung diseases, can develop a severe acute respiratory syndrome (SARS), requiring mechanical ventilation and admission to an intensive care unit (ICU) [8][9]. The high prevalence of obesity and T2D around the world reveals a huge public health demand to combat COVID-19 infection. Currently, obesity affects more than 650 million people and 463 million has T2D worldwide [10][11]. Both diseases share a common feature, augmented adiposity associated with a chronic systemic low-grade inflammation which induce dysregulation of the immune system and increasing susceptibility to develop infections [8][12][13][14][15][16]. The pre-existing chronic inflammation in obese and T2D patients with the augmented inflammatory response against the viral infection seems to increase the susceptibility of these patients for developing severe COVID-19.
2. How Immune System Fight against Virus
To know how viruses interact with the host immune system is essential for understanding the virulence, pathogenesis, and disease outcomes.
2.1. Role of Macrophages and Dendritic Cells against Virus
The innate immune response is the host’s first line of defense to prevent and eliminate infection of the invading virus. The early natural immune activation also plays an important role in stimulating the adaptive immune response [17]. When the virus enters the host cells, a signaling cascade is initiated which triggers several mechanism of innate defense such us inflammation followed by a repair phase when the antigen is cleared [18]. The capacity of the innate immune cells to sense danger is essential for well-regulated immune responses. Macrophages (MP) and Dendritic cells (DCs) are the first line cells of the immune system. Activation of MP by viral agents induce these cells to response secreting inflammatory cytokines, which activate other immune cells generating a positive feedback loop of inflammation. However, the exaggerated inflammatory response can induce pyroptosis (a type of cell death) which may ultimately lead to virus induced immune pathology [19]. MP also secrete chemokines to recruit other immune cells to the site of infection to eliminate the pathogen. Once the antigen is eliminated, MP secrete molecules to balance the immune response to induce tissue regeneration. DCs play a key role in both innate and adaptive immune responses to viral pathogens [20]. There are two main types of DCs, plasmacytoid (pDC) and myeloid (mDC) cells. pDC produce large amounts of type I IFN, which can induce direct antiviral state [21]. mDCs are specialized antigen-presenting cells (APC) which processes the endogenous and exogenous antigens for presentation by major histocompatibility complex (MHC) molecules to T cells through the T-cell antigen receptors (TCR) [18]. MHC-II molecules are expressed on epithelial cells and immune cells such as B cells, monocytes, macrophages and dendritic cells instead MHC class I molecules are expressed in almost all nucleated cells [22]. MHC-I and MHC-II present protein antigen fragments to CD8+ and CD4+ T cells, respectively. Thus, the pattern expression of MHC molecules directs T cells to interact with exactly the right kind of cells. Once mDCs encounters an antigen, they become activated to produce alarm signals in the form of inflammatory cytokines and costimulatory molecules needed to stimulate host cells [18][23][24]. Another important component of the innate immune system key to fight against virus, are the pattern recognition receptors (PRRs) (e.g., Toll-like, NOD and RIG-I receptors), which are engaged to detect viral RNA or DNA to induce pro-inflammatory cytokines and type I interferons (IFNs) in the infected and other immune cells to eliminate the invading virus [25][26][27]. If the virus cannot be eliminated by natural immune responses, the adaptive immune system uses innate signals to help initiate and enhance its activation [28].
2.2. Role of CD8+ T Cells against Virus
To become an effector cell, a naïve CD8+ T cell must receive 3 signals to trigger efficient immune responses [29][30]. Once naïve CD8+ T cells recognize foreign antigens presented to them, through the major histocompatibility complex class I (MHC-I); they initiate adaptive immune responses, specifically against these antigens using a unique antigen-binding site in the CDR3 domain of the TCR receptor [31][32][33]. This antigenic stimulation (signal 1) induces T cell division to give rise to a clone of cells with identical specificities [31][34][35]. The second stimulation (signal 2) is generated through the engagement of costimulatory molecules, namely B7-1 (CD80)/B7-2 (CD86) in APCs and CD28 in T cells [36][37][38]. In case, B7 molecules recognize the cytotoxic T lymphocyte antigen 4 (CTLA-4), instead of CD28, the opposite functional effect occurs, thus inducing inhibition [39][40]. Another costimulatory molecule, crucial for CD8+ CTLs activation and memory formation is CD40 [41][42]. The interaction of CD40-expressing CD8+ T cells and CD40L of CD4+ T helper (Th) cells induce activation of CD8+ T cells directly [26][42][43]. Thus, CD8+ T cell responses to all antigens require CD40 signaling. Third, CD8+ T cells receive specific cytokine signals (signal 3), which further enhance, modify, and skew the responding effect [34][35][38]. The described stimulatory signals take place in the formed immunologic synapse, surrounded also by rings of engaged accessory integrin molecules [44][45]. If one of the signals is not present, tolerance occur instead [46]. Once activated, CD8+ T cells become cytotoxic cells (CTL) which are critical to fight against virus. CTLs exercise their cytotoxicity mediated function through the release of granzymes and perforins, FAS ligand/tumor necrosis factor inducing pathways to kill the infected target cell by apoptosis [18][47][48]. After CTLs perform their “effector” function, the cell population contracts and the remaining cells constitute a long-lived memory CD8+ T-cell pool that protects from secondary infection [49][50][51]. Virus can directly infect DC, which theoretically allow direct presentation of viral antigens to CD8+ T cells, but many viruses target other cells than DC, for example lung epithelial cells and thus the host depends on the cross-presentation of viral antigens by DC to activate virus-specific CD8+ T cells [52][53]. The DCs subsets involved in cross-presentation differ depending on the site of infection and the inflammatory environment [54]. Only when DCs are activated, they can trigger effective CD8+ T-cell responses inducing activation and memory formation [55].
2.3. Role of CD4+ T Cells against Virus
CD4+ T cells also have a crucial role in antiviral immune responses. Evidence suggests that in cells where viruses replicate (e.g., lung epithelial cells), a number of viral proteins found in the cytoplasm, can load recycled MHC class II molecules in early endosomes or phagosomes for presentation to naïve CD4+ T cells [56]. However, as defense mechanism, virus can block the induction of class II genes and interfere with the loading of viral peptides [57]. The presentation of viral antigens by APCs (DCs, MP, and B cells) through MHC-II induce activation of naïve CD4+ T cells, which originate T helper (Th) cells with different phenotypes and functions (e.g., Th1, Th2, Th17). The dominant cytokine environment, costimulatory molecules and type of antigen presented, determines the polarized CD4+ T helper formation. Activated CD4+ T cells can help B cells to generate stronger and longer-lived antibody responses, which neutralizing function are key in the defense against virus. CD4+ T cells also can help CD8+ T cell to induce its expansion during a primary immune response facilitating the generation of virus-specific memory CD8+ T cell populations [58]. Additionally, CD4+ Th1-stimulatory cytokines like IL-18 and IL-12 stimulates natural killer cells (NK) to exert their cytotoxicity activity [59][60][61]. Apart from their helper function, evidence from animal models suggests that effector CD4+ T cells can contribute directly to viral clearance secreting cytokines with antiviral activities, and functioning like cytotoxic killing cells [62][63][64]. Finally, it is described that once formed, memory CD4+ T cells have enhanced helper and effector activity that allow them rapidly trigger immune defense mechanisms [56][58].
2.4. Role of B Cells against Virus
Conventional B cells are key players in the generation of specific and long-lasting antibodies against virus and require the collaboration of other cells [65]. Naïve B-cell activation occurs after the viral encounter, which can occur directly via antigen recognition through B cell receptor (BCR) or through MHC dendritic cell antigen presentation to B cells in the lymph nodes [66][67]. Thus, MHC class II molecules play an important role in the interaction with B cells to produce antibodies [58][59][68]. Following antigen presentation, B cells activates either extra follicular or germinal centers. Extra follicular B-cell responses will generate short-lived and no memory B cells therefore, much of the early-induced antiviral antibody responses, does not seem to contribute to the long-term response to the virus [65]. On the other hand, germinal center (GC) responses generate high-affinity antibody-secreting plasma cells and long-term memory B cells that ensure sustained immune protection, and rapid recall responses against previously encountered foreign antigens [65]. GC responses are the basis of T cell dependent collaboration, where somatic hypermutation and affinity maturation of B cells occurs. Interestingly, it is described that despite the clearance of viral infection, the germinal centers can persist for many months [69]. The first immunoglobulin produced by plasma cells is the IgM, which has low affinity and a relatively short half-live (around 7 days). IgM secretion peaks appears early and then disappears within a few weeks of infection [69]. If the antigen activated B cells receive collaboration of T helper cells via CD40 molecule, they undergo antibody class switching in the constant region of the immunoglobulin heavy chain to produce antibodies of different isotypes or subtypes (IgG, IgA, or IgE) [70][71]. Those antibodies will conserve the antigenic specificity, but their function will differ depending of its localization. IgG is the principal isotype in the blood and extracellular fluids, whereas IgA is the principal isotype in secretions (intestinal and respiratory tract) [67]. IgG is always monomeric, its secretion peaks around 14 days after infection and decreases over time but do not usually return to baseline levels [65]. Secreted IgG has a central importance to protect against viruses since efficiently neutralize viral antigens [72][73]. In addition, it can opsonize viral antigens for engulfment by phagocytes, participate in antibody-dependent cellular cytotoxicity (ADCC) by NK cells, and activates the complement system to mediated lysis of infected cells. IgA form dimers, is a less potent opsonin, and is a weak activator of complement [74][75]. The affinity of the antigen-binding sites for their antigen is critical for the effectiveness of these antibodies against viral pathogens [65]. Although T and B-lymphocytes function differs from those of innate immunity, the functional pathways largely overlap.
Overall, the innate and adaptive arms of the immune response should be viewed as complementary and cooperating. Its proper function is required for an effective virus clearance and inappropriate activation. In many viral infectious diseases, the principal pathological aspects are not related to the direct action of an aggressor agent, but instead by the “virus’ cytopathic effect” or through “immune cytotoxicity”.
3. Immune Alterations in Obesity and T2D
Metabolic diseases such as obesity and T2D share a common feature, augmented adiposity associated with a chronic systemic low-grade inflammation [12][13] which promotes the abnormal production of pro-inflammatory cytokines and the impairment of the immune response and host defense [8]. Adipose tissue is an endocrine organ capable of secreting a diversity of cytokines and adipokines that are involved in the regulation of inflammation and homeostasis [76]. Healthy adipocytes are sensitive to insulin, which is critical for the uptake of glucose and maintenance of blood glucose levels. In obese subjects, adipocytes increase in number and size. This phenomenon is accompanied by an insufficient vascularization of the tissue resulting in hypoxia, triggering apoptosis and/or necrosis as well as elevated secretion of more inflammatory cytokines, adipokines, and chemokines that induce a vast infiltration of immune cells contributing to lipolysis, promoting inflammation and insulin resistance. This metabolic impairment induces the initiation or aggravation of T2D [12]. All these factors generate a low-grade inflammatory microenvironment that incites the recruitment of mast cells, neutrophils, T-cells, B-cells, and the polarization of inflammatory macrophages towards the M1 phenotype is favored in adipose tissue of obese subjects, while maintaining or even reducing the number of regulatory T cells (Treg), T helper cells type 2 (Th2), and M2 macrophages [77]. As a result, the balance shifts from a regulatory anti-inflammatory immune state characterized for the secretion of cytokines IL-4, IL-5, IL-10, IL-13, and IL-33 to an exceedingly inflammatory state secreting multiple proinflammatory cytokines, such as IL-6, IL-8, TNF-α, IL-1β, and chemokine (C-C motif) ligand 2 (CCL2), contributing to maintain a chronic systemic inflammation [6][78]. Additionally, the elevated levels of IL-6 provoke an acute-phase response which increases ferritin, C-Reactive Protein (CRP) and D-dimer [79]. The action of pro-inflammatory cytokines in immune cells can trigger signaling pathways that converge at the NF-κB and MAPK activation, resulting in the generation of intracellular inflammatory cytokine production (IL-1β, IL-6, and TNF-α) and apoptosis induction, respectively. In addition, the increased ROS production induced by excess of fatty acids and high glucose triggers the activation of NLR family pyrin domain containing 3 (NLRP3) inflammasome that, through caspasse-1 activates IL-1β form. Overall, the activation of intracellular pathways accompanied by adipose inflammation increases the production of pro-inflammatory cytokines and promotes the infiltration of more pro-inflammatory M1 macrophages. Additionally, a hallmark in obesity and T2D are the senescent immune cells which exhibit a senescence-associated secretory phenotype (SASP) characterized by secreting a huge quantity of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, IL-18, CCL-2, and TNF-α) [80][81][82]. SASP cells have enhanced mitochondrial apoptosis switching from apoptosis to pyroptosis, through a high mitogen-activated protein kinases (MAPKs) activity which drive IL-1 production (leading to auto-inflammation) and reduced IFN type 1 production, leading to a deficiency to combat viral infections [83] (see Figure 1). The inflammatory cytokines also induce neuro-immune-endocrine interactions affecting the body’s response to stress. Catecholamines secreted by the sympathetic nervous system (SNS) and the adrenal glands are key regulators of metabolism and innate responses which are affected in obesity [84][85]. Animal models of metabolic syndrome have demonstrated the feedback mechanism between inflammatory cytokines and noradrenaline (NA) and the altered phagocytic and microbicidal capacity of macrophages, which caused a greater susceptibility to infections [86][87]. In fact, recent studies have proposed a novel adrenergic regulation of inflammation, via activation of β2-adrenergic receptors, highly expressed in immune cells, to modulate the inflammatory phenotype and activity profile of these cells [85][88].
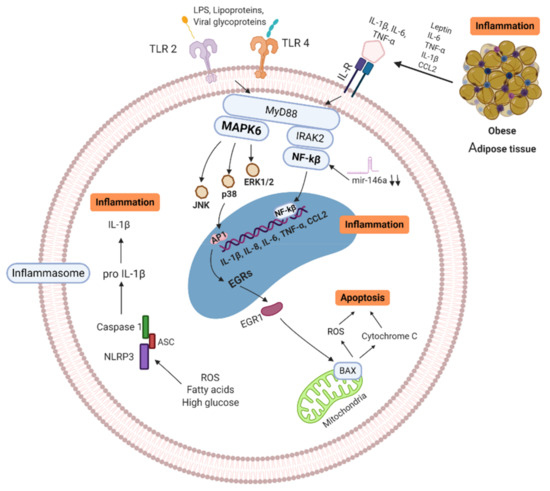
Figure 1. Signaling pathways of obese/T2D immune cell (MNC/MP). Senescence-associated secretory phenotype (SASP) cells secrete high quantity of pro-inflammatory cytokines, which trigger enhanced mitochondrial apoptosis through high mitogen-activated protein kinases (MAPKs). ROS production, free fatty acids and high glucose induce activation of NLRP3 inflammasome, activating IL-1β, leading to further inflammation. In addition, Pattern Recognition Receptors (PRR) and Interleukin Receptors (ILR) signaling through NF-κB and MAPKs pathways, resulting in more inflammatory cytokine production (IL-1β, IL-6, IL-8, IL-18, CCL-2, and TNF-α). Activation of these pathways increases the production of pro-inflammatory cytokines and promotes the infiltration of more pro-inflammatory M1 macrophages in tissues. MAPK: Mitogen-activated protein kinase, IL: Interleukins, ILR: Interleukins receptor, TNF: Tumor necrosis factor, BAX: BCL2-associated X genes, JNK: c-Jun N-terminal kinase, IRAK-2: Interleukin-1 receptor-associated kinase 2, EGRs: Early response genes, NF-κB: nuclear factor kappa B, TLR: Toll-like receptor, CCL2: C-C motif chemokine ligand 2, ROS: Reactive oxygen species, mir146a: microRNA-146a, NLRP3: NLR family pyrin domain containing 3, ASC: Adaptor protein. Created with BioRender.com.
Besides, important adipokines involved in the inflammatory process are leptin and adiponectin. Leptin is a lipostatic hormone whose main function is to regulate body weight by transmitting signals of satiety to the central nervous system and energy homeostasis [89][90][91]. Evidence shows that leptin is involved in glucose metabolism, innate immune reactions and acute inflammation. The concentration of leptin in blood in obese patients with dyslipidemia is high, causing signaling alteration, promoting the development of insulin resistance and T2D [92][93]. In mice, it has been described an increase in leptin is associated with inflammatory markers in obese individuals [94], and in humans production of cytokines can be induce when leptin is administered exogenously [95]. Adiponectin is a protein whose function is to increase fat oxidation, producing a reduction in the concentration of fatty acids, increasing insulin sensitivity [90][96]. It is described that adiponectin levels decrease in obesity, insulin resistance, T2D and cardiovascular diseases [97]. In addition, alteration in the relationship between leptin and adiponectin ratio has been shown to be associated with insulin resistance [98][99], and in the development of atherosclerosis [100]. Consequently, leptin and adiponectin play an important role in the regulation of cardiovascular and metabolic homeostasis [91][96][101].
MicroRNAs (miRNA) are other important biomarkers that play a relevant role in obesity/T2D [102][103][104]. MiRNAs regulate and modulate the expression of a large number of protein-coding genes [105]. Several studies indicate that miRNA-146a and miRNA-155 are involved in the regulation of inflammatory processes [106][107]. For example, miRNA-155 can promote the activation of the LPS/TNF pathway, thus contributing to the activation of the inflammatory response [108][109]. MiRNA-146a block the nuclear factor kappa B (NF-κB) activation induced by TNF-α and toll-like receptor ligands, therefore its function is crucial in the prevention of excessive immune response [110]. Baldeón L et al. showed that the expression levels of miR-146a in CD14+ in T2D monocytes, was down-regulated when compared to healthy controls. The decreased expression of miR-146a in immune cells, correlates negatively with inflammatory cytokines, thus acting as a negative regulator during the activation of immune responses [111].
Adaptive immune cells (T cells) may play a significant role in the propagation of adipose tissue inflammation in obesity. T cells from obese/T2D patients express lower levels of costimulatory molecules (CD69, CD28, CD40 ligand), and interleukin-12 receptor, as well as, produce lower levels of interferon-γ and granzyme B, compared to healthy individuals [112][113][114]. It was shown in diet-induced obesity mice model that CD8+ T cells infiltrate into fat pads before macrophage infiltration. Additionally, CD8+ T cell depletion with anti CD8+ antibodies, resulted in reduced M1 macrophage infiltrations and decreased inflammatory mediators in adipose tissue, ameliorating insulin resistance and glucose tolerance [115]. Conversely, Winer et al. found that the progression of obesity-associated metabolic abnormalities is under the pathophysiological control of CD4+ T cells. Reconstitution of CD4+ T cells, but not CD8+ T cells, in lymphocyte-free obese Rag1-null mice improved glucose tolerance, enhanced insulin sensitivity, and reduced weight gain [116]. Furthermore, there is evidence that regulatory T cells (Treg) are key regulatory cells in adipose tissue to provide anti-inflammatory signals that block adipose tissue inflammation. Treg cells normally account for 5%–20% of the CD4+ compartment but are thought to be one of the body’s most crucial defenses against inappropriate immune responses [117][118][119]. For example, Feuerer et al. demonstrated that when most of the Treg cells were ablated in fat tissue, proinflammatory transcripts were strongly expressed, suggesting the important anti-inflammatory properties of Treg in metabolic processes [120].
B-lymphocytes have been shown to play a central role in the development of insulin resistance and glucose intolerance by activating CD4+ Th1 and Th17 cells and releasing pathogenic antibodies [121][122]. Epigenetic modifications such as DNA methylation play and important role in the significant increase of B cells proliferation which in turn influence the proliferation of Th17 and the production of proinflammatory cytokines [114][123]. For example, Ip et al. show that in vitroTh17 cells decreased upon depletion of CD19+ cells from PBMCs of T2D patients [124]. In addition, in an animal model it was shown that the lack of mature B cells improved glucose tolerance in HFD mice [114]. Interestingly, posttranscriptional modifications in which various glycosyltransferases are involved are well described to occur in inflammatory disease such as obesity and T2D [125][126]. Wu et al. described that IgG glycosylation profiles were associated with T2D, arguing that glycan score can be used as novel indicator for diabetes and inflammatory status [127].
In conclusion, metabolic disorders such as obesity and T2D, seems to alter the balance of innate and adaptive responses, demonstrated by senescence, exaggerated cytokine inflammation, poor chemotaxis, impaired phagocytosis, glycation of circulating immunoglobulins, and abnormalities in the number and function of CD4+ T and CD8+ T lymphocytes and B lymphocytes.
References
- Chehrehgosha, M. The Unpreparedness of the Healthcare System for the Management of COVID-19 Pandemic Leading to the Mistreatment of the Elderly: A Newly Emerging Moral Dilemma. J. Nutr. Heal. Aging 2020.
- Gallo Marin, B.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 severity: A literature review. Rev. Med. Virol. 2020, e2146.
- Nieman, D.C. Coronavirus disease-2019: A tocsin to our aging, unfit, corpulent, and immunodeficient society. J. Sport Health Sci. 2020.
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020.
- Johns Hopkins University. COVID-19 Map—Johns Hopkins Coronavirus Resource Center; Johns Hopkins Coronavirus Resource Center: Baltimore, MA, USA, 2020.
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648.
- Deng, Y.; Liu, W.; Liu, K.; Fang, Y.Y.; Shang, J.; Zhou, L.; Wang, K.; Leng, F.; Wei, S.; Chen, L.; et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: A retrospective study. Chin. Med. J. 2020, 133, 1261–1267.
- Chiappetta, S.; Sharma, A.M.; Bottino, V.; Stier, C. COVID-19 and the role of chronic inflammation in patients with obesity. Int. J. Obes. 2020.
- Yan, Y.; Yang, Y.; Wang, F.; Ren, H.; Zhang, S.; Shi, X.; Yu, X.; Dong, K. Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res. Care 2020.
- Vaamonde, J.G.; Álvarez-Món, M.A. Obesity and overweight. Medicine 2020.
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; IDF: Brussels, Belgium, 2019; ISBN 9782930229874.
- Muniyappa, R.; Gubbi, S. COVID-19 pandemic, coronaviruses, and diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2020.
- Ryan, P.M.D.; Caplice, N.M. Is Adipose Tissue a Reservoir for Viral Spread, Immune Activation, and Cytokine Amplification in Coronavirus Disease 2019? Obesity 2020, 28, 1191–1194.
- Daryabor, G.; Atashzar, M.R.; Kabelitz, D.; Meri, S.; Kalantar, K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front. Immunol. 2020.
- de Lucena, T.M.C.; da Silva Santos, A.F.; de Lima, B.R.; de Albuquerque Borborema, M.E.; de Azevêdo Silva, J. Mechanism of inflammatory response in associated comorbidities in COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020.
- Dai, S.P.; Zhao, X.; Wu, J.H. Effects of Comorbidities on the Elderly Patients with COVID-19: Clinical Characteristics of Elderly Patients Infected with COVID-19 from Sichuan, China. J. Nutr. Heal. Aging 2020.
- Cruz-Tapias, P.; Castiblanco, J.; Correa, N.E.; Montoya-Ortíz, G. AUTOIMMUNITY From Bench to Bedside; El Rosario University Press: Bogota, Colombia, 2013; ISBN 978-958-738-376-8.
- Janeway, C.A. How the immune system protects the host from infection. Microbes Infect. 2001.
- Frieman, M.; Heise, M.; Baric, R. SARS coronavirus and innate immunity. Virus Res. 2008.
- Lee, H.K.; Iwasaki, A. Innate control of adaptive immunity: Dendritic cells and beyond. Semin. Immunol. 2007.
- Mathan, T.S.M.; Figdor, C.G.; Buschow, S.I. Human plasmacytoid dendritic cells: From molecules to intercellular communication network. Front. Immunol. 2013.
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016.
- Kotsias, F.; Cebrian, I.; Alloatti, A. Antigen processing and presentation. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128183519.
- Zuniga, E.I.; McGavern, D.B.; Oldstone, M.B.A. Antigen Presentation. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008.
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820.
- Castellino, F.; Germain, R.N. Cooperation between CD4+ and CD8+ T cells: When, where, and how. Annu. Rev. Immunol. 2006.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010.
- Hansson, G.K.; Libby, P.; Schönbeck, U.; Yan, Z.Q. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 2002.
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010.
- Garcia, K.C.; Teyton, L.; Wilson, I.A. Structural basis of T cell recognition. Annu. Rev. Immunol. 1999.
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988.
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013.
- Lanzavecchia, A.; Sallusto, F. Antigen decoding by T lymphocytes: From synapses to fate determination. Nat. Immunol. 2001.
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004.
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002.
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996.
- Condotta, S.A.; Richer, M.J. The immune battlefield: The impact of inflammatory cytokines on CD8+T-cell immunity. PLoS Pathog. 2017, 13, e1006618.
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T ceils to stimulation. J. Exp. Med. 1995, 182, 459–465.
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182.
- Van Kooten, G.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17.
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40–CD40L axis in immunity against infection and cancer. ImmunoTargets Ther. 2018.
- Xiang, J.; Huang, H.; Liu, Y. A New Dynamic Model of CD8 + T Effector Cell Responses via CD4 + T Helper-Antigen-Presenting Cells. J. Immunol. 2005.
- Zhu, Y.; Yao, S.; Chen, L. Cell Surface Signaling Molecules in the Control of Immune Responses: A Tide Model. Immunity 2011.
- Ahmed, K.A.; Xiang, J. Mechanisms of cellular communication through intercellular protein transfer. J. Cell. Mol. Med. 2011.
- Quezada, S.A.; Jarvinen, L.Z.; Lind, E.F.; Noelle, R.J. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004.
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161–168.
- Harty, J.T.; Tvinnereim, A.R.; White, D.W. Cd8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 2000.
- Bourgeois, C.; Rocha, B.; Tanchot, C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 2002.
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011.
- Schluns, K.S.; Lefrançois, L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003.
- Embgenbroich, M.; Burgdorf, S. Current concepts of antigen cross-presentation. Front. Immunol. 2018.
- van Montfoort, N.; van der Aa, E.; Woltman, A.M. Understanding MHC class I presentation of viral antigens by human dendritic cells as a basis for rational design of therapeutic vaccines. Front. Immunol. 2014.
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012.
- Schönbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001.
- Sant, A.J.; McMichael, A. Revealing the role of CD4+ T cells in viral immunity. J. Exp. Med. 2012.
- Hegde, N.R.; Chevalier, M.S.; Johnson, D.C. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 2003.
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4 + T cells in immunity to viruses. Nat. Rev. Immunol. 2012.
- Machado, P.R.L.; Carvalho, L.; Araújo, M.I.A.S.; Carvalho, E.M. Immune response mechanisms to infections. An. Bras. Dermatol. 2004.
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407.
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001.
- Appay, V. The physiological role of cytotoxic CD4+ T-cells: The holy grail? Clin. Exp. Immunol. 2004.
- Casazza, J.P.; Betts, M.R.; Price, D.A.; Precopio, M.L.; Ruff, L.E.; Brenchley, J.M.; Hill, B.J.; Roederer, M.; Douek, D.C.; Koup, R.A. Acquisition of direct antiviral effector functions by CMV-specific CD4 + T lymphocytes with cellular maturation. J. Exp. Med. 2006.
- Soghoian, D.Z.; Jessen, H.; Flanders, M.; Sierra-Davidson, K.; Cutler, S.; Pertel, T.; Ranasinghe, S.; Lindqvist, M.; Davis, I.; Lane, K.; et al. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 2012.
- Baumgarth, N. How specific is too specific? B-cell responses to viral infections reveal the importance of breadth over depth. Immunol. Rev. 2013.
- Gonzalez, S.F.; Lukacs-Kornek, V.; Kuligowski, M.P.; Pitcher, L.A.; Degn, S.E.; Kim, Y.A.; Cloninger, M.J.; Martinez-Pomares, L.; Gordon, S.; Turley, S.J.; et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 2010.
- Lam, J.H.; Baumgarth, N. The Multifaceted B Cell Response to Influenza Virus. J. Immunol. 2019.
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015.
- Rothaeusler, K.; Baumgarth, N. B-cell fate decisions following influenza virus infection. Eur. J. Immunol. 2010.
- Basso, K.; Klein, U.; Niu, H.; Stolovitzky, G.A.; Tu, Y.; Califano, A.; Cattoretti, G.; Dalla-Favera, R. Tracking CD40 signaling during germinal center development. Blood 2004.
- MacLennan, I.C.M. Germinal centers. Annu. Rev. Immunol. 1994.
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Seaman, M.S.; Velinzon, K.; Pietzsch, J.; Ott, R.G.; Anthony, R.M.; Zebroski, H.; Hurley, A.; et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009.
- Yu, X.; Tsibane, T.; McGraw, P.A.; House, F.S.; Keefer, C.J.; Hicar, M.D.; Tumpey, T.M.; Pappas, C.; Perrone, L.A.; Martinez, O.; et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 2008.
- Dörner, T.; Radbruch, A. Antibodies and B Cell Memory in Viral Immunity. Immunity 2007.
- Hooijkaas, H.; Benner, R.; Pleasants, J.R.; Wostmann, B.S. Isotypes and specificities of immunoglobulins produced by germ-free mice fed chemically defined ultrafiltered “antigen-free” diet. Eur. J. Immunol. 1984.
- Goossens, G.H.; Dicker, D.; Farpour-Lambert, N.J.; Fruhbeck, G.; Mullerova, D.; Woodward, E.; Holm, J.C. Obesity and COVID-19: A Perspective from the European Association for the Study of Obesity on Immunological Perturbations, Therapeutic Challenges, and Opportunities in Obesity. Obes. Facts 2020, 13, 439–452.
- Kang, Z.; Luo, S.; Gui, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Zhou, Q.; Wang, Q.; Hu, Y.; Fan, H.; et al. Obesity is a potential risk factor contributing to clinical manifestations of COVID-19. Int. J. Obes. 2020.
- Ritter, A.; Kreis, N.N.; Louwen, F.; Yuan, J. Obesity and covid-19: Molecular mechanisms linking both pandemics. Int. J. Mol. Sci. 2020, 21, 5793.
- Méry, G.; Epaulard, O.; Borel, A.L.; Toussaint, B.; Le Gouellec, A. COVID-19: Underlying Adipokine Storm and Angiotensin 1-7 Umbrella. Front. Immunol. 2020, 11, 1–10.
- Stojanovic, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020.
- Chiu, Y.-L.; Tsai, W.-C.; Hung, R.-W.; Chen, I.-Y.; Shu, K.-H.; Pan, S.-Y.; Yang, F.-J.; Ting, T.-T.; Jiang, J.-Y.; Peng, Y.-S.; et al. Emergence of T cell immunosenescence in diabetic chronic kidney disease. Immun. Ageing 2020.
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016.
- Beumer, W.; Drexhage, R.C.; De Wit, H.; Versnel, M.A.; Drexhage, H.A.; Cohen, D. Increased level of serum cytokines, chemokines and adipokines in patients with schizophrenia is associated with disease and metabolic syndrome. Psychoneuroendocrinology 2012.
- Ortega, E.; Gálvez, I.; Martín-Cordero, L. Adrenergic Regulation of Macrophage-Mediated Innate/Inflammatory Responses in Obesity and Exercise in this Condition: Role of β2 Adrenergic Receptors. Endocr. Metab. Immune Disord. Drug Targets 2019.
- Martín-Cordero, L.; Gálvez, I.; Hinchado, M.D.; Ortega, E. Influence of Obesity and Exercise on β2-Adrenergic-Mediated Anti-Inflammatory Effects in Peritoneal Murine Macrophages. Biomedicines 2020, 8, 556.
- Martín-Cordero, L.; García, J.J.; Hinchado, M.D.; Ortega, E. The interleukin-6 and noradrenaline mediated inflammation-stress feedback mechanism is dysregulated in metabolic syndrome: Effect of exercise. Cardiovasc. Diabetol. 2011.
- García, J.J.; del Carmen Sáez, M.; De la Fuente, M.; Ortega, E. Regulation of phagocytic process of macrophages by noradrenaline and its end metabolite 4-hydroxy-3-metoxyphenyl-glycol. Role of α- and β-adrenoreceptors. Mol. Cell. Biochem. 2003.
- Martín-Cordero, L.; Gálvez, I.; Hinchado, M.D.; Ortega, E. β2 Adrenergic Regulation of the Phagocytic and Microbicide Capacity of Macrophages from Obese and Lean Mice: Effects of Exercise. Nutrients 2019, 11, 2721.
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599.
- Aly, O.; Zaki, H.H.; Herzalla, M.R.; Fathy, A.; Raafat, N.; Hafez, M.M. Gene polymorphisms of Patatin-like phospholipase domain containing 3 (PNPLA3), adiponectin, leptin in diabetic obese patients. PLoS ONE 2020, 15, e0234465.
- Inoue, M.; Maehata, E.; Yano, M.; Taniyama, M.; Suzuki, S. Correlation between the adiponectin-leptin ratio and parameters of insulin resistance in patients with type 2 diabetes. Metabolism 2005, 54, 281–286.
- Bidulescu, A.; Dinh, P.C.; Sarwary, S.; Forsyth, E.; Luetke, M.C.; King, D.B.; Liu, J.; Davis, S.K.; Correa, A. Associations of leptin and adiponectin with incident type 2 diabetes and interactions among African Americans: The Jackson heart study. BMC Endocr. Disord. 2020, 20.
- Thorand, B.; Zierer, A.; Baumert, J.; Meisinger, C.; Herder, C.; Koenig, W. Associations between leptin and the leptin/adiponectin ratio and incident Type 2 diabetes in middle-aged men and women: Results from the MONICA/KORA Augsburg Study 1984–2002. Diabet. Med. 2010, 27, 1004–1011.
- Van Dielen, F.M.H.; Van’t Veer, C.; Schols, A.M.; Soeters, P.B.; Buurman, W.A.; Greve, J.W.M. Increased leptin concentrations correlate with increased concentrations of inflammatory markers in morbidly obese individuals. Int. J. Obes. 2001, 25, 1759–1766.
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9.
- Satoh, N.; Naruse, M.; Usui, T.; Tagami, T.; Suganami, T.; Yamada, K.; Kuzuya, H.; Shimatsu, A.; Ogawa, Y. Leptin-to-adiponectin ratio as a potential atherogenic index in obese type 2 diabetic patients. Diabetes Care 2004, 27, 2488–2490.
- Al-Hamodi, Z.; Al-Habori, M.; Al-Meeri, A.; Saif-Ali, R. Association of adipokines, leptin/adiponectin ratio and C-reactive protein with obesity and type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2014, 6.
- Oda, N.; Imamura, S.; Fujita, T.; Uchida, Y.; Inagaki, K.; Kakizawa, H.; Hayakawa, N.; Suzuki, A.; Takeda, J.; Horikawa, Y.; et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism 2008, 57, 268–273.
- Finucane, F.M.; Luan, J.; Wareham, N.J.; Sharp, S.J.; O’Rahilly, S.; Balkau, B.; Flyvbjerg, A.; Walker, M.; Højlund, K.; Nolan, J.J.; et al. Correlation of the leptin: Adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia 2009, 52, 2345–2349.
- Kotani, K.; Sakane, N.; Saiga, K.; Kurozawa, Y. Leptin: Adiponectin ratio as an atherosclerotic index in patients with type 2 diabetes: Relationship of the index to carotid intima-media thickness. Diabetologia 2005, 48, 2684–2686.
- Matsubara, M.; Maruoka, S.; Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 2002, 147, 173–180.
- Kolfschoten, I.G.M.; Roggli, E.; Nesca, V.; Regazzi, R. Role and therapeutic potential of microRNAs in diabetes. Diabetes, Obes. Metab. 2009, 11, 118–129.
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859.
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019.
- DP, B. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function.pdf. Cell 2004, 116, 281–297.
- McClelland, A.D.; Kantharidis, P. microRNA in the development of diabetic complications. Clin. Sci. 2014, 126, 95–110.
- Roos, J.; Enlund, E.; Funcke, J.B.; Tews, D.; Holzmann, K.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P. MiR-146a-mediated suppression of the inflammatory response in human adipocytes. Sci. Rep. 2016, 6, 1–11.
- Ge, Q.; Brichard, S.; Yi, X.; Li, Q. MicroRNAs as a new mechanism regulating adipose tissue inflammation in obesity and as a novel therapeutic strategy in the metabolic syndrome. J. Immunol. Res. 2014, 2014, 987285.
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b Levels following Lipopolysaccharide/TNF-α Stimulation and Their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089.
- Sonkoly, E.; Ståhle, M.; Pivarcsi, A. MicroRNAs and immunity: Novel players in the regulation of normal immune function and inflammation. Semin. Cancer Biol. 2008, 18, 131–140.
- Baldeón R, L.; Weigelt, K.; de Wit, H.; Ozcan, B.; van Oudenaren, A.; Sempértegui, F.; Sijbrands, E.; Grosse, L.; Freire, W.; Drexhage, H.A.; et al. Decreased serum level of miR-146a as sign of chronic inflammation in type 2 diabetic patients. PLoS ONE 2014, 9, e115209.
- Delhanty, P.J.D.; Huisman, M.; Baldeon-rojas, L.Y.; Van Den Berge, I.; Grefhorst, A.; Abribat, T.; Leenen, P.J.M.; Themmen, A.P.N.; Van Der Lely, A. Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. FASEB J. 2013, 27, 1690–1700.
- Xia, C.; Rao, X.; Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J. Diabetes Res. 2017.
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013.
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009.
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009.
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011.
- Zheng, Y.; Chaudhry, A.; Kas, A.; DeRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature 2009.
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008.
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009.
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127.
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011.
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barrès, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 2014.
- Ip, B.; Cilfone, N.A.; Belkina, A.C.; Defuria, J.; Jagannathan-Bogdan, M.; Zhu, M.; Kuchibhatla, R.; McDonnell, M.E.; Xiao, Q.; Kepler, T.B.; et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity 2016.
- Shade, K.-T.; Anthony, R. Antibody Glycosylation and Inflammation. Antibodies 2013, 2, 392–414.
- Alter, G.; Ottenhoff, T.H.M.; Joosten, S.A. Antibody glycosylation in inflammation, disease and vaccination. Semin. Immunol. 2018.
- Wu, Z.; Li, H.; Liu, D.; Tao, L.; Zhang, J.; Liang, B.; Liu, X.; Wang, X.; Li, X.; Wang, Y.; et al. IgG Glycosylation Profile and the Glycan Score Are Associated with Type 2 Diabetes in Independent Chinese Populations: A Case-Control Study. J. Diabetes Res. 2020.



