 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Batie | + 6016 word(s) | 6016 | 2021-02-18 12:30:24 | | | |
| 2 | Lily Guo | Meta information modification | 6016 | 2021-03-04 03:34:11 | | | | |
| 3 | Lily Guo | + 2 word(s) | 6018 | 2021-03-04 04:24:37 | | |
Video Upload Options
Hypoxia—reduction in oxygen availability—plays key roles in both physiological and pathological processes. Given the importance of oxygen for cell and organism viability, mechanisms to sense and respond to hypoxia are in place. A variety of enzymes utilise molecular oxygen, but of particular importance to oxygen sensing are the 2-oxoglutarate (2-OG) dependent dioxygenases (2-OGDs). Of these, Prolyl-hydroxylases have long been recognised to control the levels and function of Hypoxia Inducible Factor (HIF), a master transcriptional regulator in hypoxia, via their hydroxylase activity. However, recent studies are revealing that dioxygenases are involved in almost all aspects of gene regulation, including chromatin organisation, transcription and translation. We highlight the relevance of HIF and 2-OGDs in the control of gene expression in response to hypoxia and their relevance to human biology and health.
1. Introduction
The importance of oxygen for energy production in multicellular organisms has been appreciated since the identification of the mechanism of oxidative phosphorylation located in the mitochondria. Reduction in oxygen availability, or hypoxia, is therefore either a danger signal or a cue for physiological processes such as development. Every cell has a different threshold for mounting a hypoxia response, although the exact mechanisms dictating this threshold are currently not well understood. Given the importance of oxygen, cells have evolved sophisticated mechanisms to sense and respond to hypoxia, in order to minimise damage, preserve energy and, when possible, adapt to the new oxygen supply normality.
The main transcription factor activated under low oxygen conditions, called Hypoxia Inducible Factor (HIF), was identified in 1992[1]. HIF is composed of a heterodimer of HIF-α (of which there are three isoforms encoded by three different genes, HIF-1α, HIF-2α and HIF3-α) and HIF-1β[2]. HIF3-α, unlike the other HIF-αs, lacks a transactivation domain, and is thought to act either as a dominant-negative or transcription-independent regulator of the hypoxic response[3]. HIFs control many genes, most of which are crucial for cell survival and adaptation to low oxygen conditions [2]. Under pathological conditions, such as cancer and altitude sickness, induction of some of these genes by the HIF transcription factors has been linked to disease progression and treatment resistance[4]. In addition, HIF can also be induced by non-oxygen dependent mechanisms, such as inflammation [5]. This is particularly relevant for human cancers, where hypoxia and inflammation often co-occur [6].
The detailed mechanism leading to the activation of HIF was unravelled in 2001[7][8], building on accumulated knowledge of the role of Von Hippel–Lindau Tumor Suppressor (VHL) in this process[9][10]. HIF-α, under normal oxygen conditions, is continually transcribed and translated, but rapidly degraded by the ubiquitin dependent proteasomal system (Figure 1). Ubiquitination is promoted by the E3-ligase composed of VHL, Ring-Box 1 (RBX1), Cullin 2 (CUL2) and Elongin B/C (ELOCB/C) [4]. VHL affinity toward HIF-α is dramatically increased by the presence of a specific post-translational modification, more precisely two separate proline hydroxylation events (Figure 1), mediated by Prolyl-Hydroxylases (PHDs). PHDs are part of the 2-oxoglutarate (2-OG) dependent dioxygenase (2-OGD) superfamily of enzymes, requiring oxygen, iron (II) and 2-OG for activity [11] Mammals possess three PHDs, PHD2 (gene name EGLN1), PHD3 (gene name EGLN3) and PHD1 (gene name EGLN2)[7].
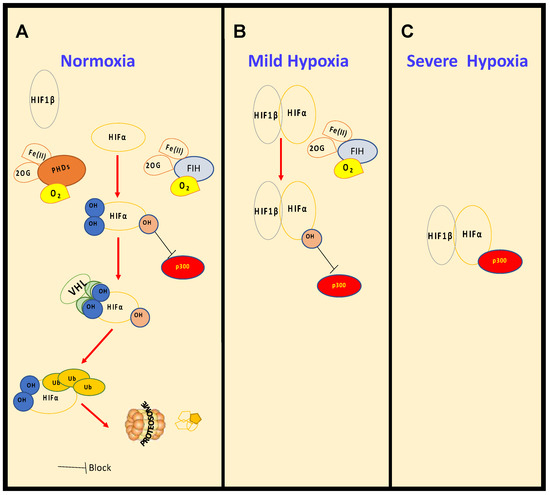
Figure 1. Regulation of HIF levels and activity in normoxia and hypoxia. Under normal oxygen conditions, (A), normoxia, HIF-α is constantly hydroxylated by PHDs and FIH. PHD-mediated hydroxylation increases binding affinity with the tumour suppressor VHL, which promotes ubiquitination and degradation by the proteosome. As oxygen levels decrease, in mild hypoxia (15–1% O2) (B), PHDs are inhibited, HIF-α is stabilised, though still hydroxylated by FIH, binds to HIF-1β and is able to induce transcription of certain target genes. With further reduction in oxygen levels, in severe hypoxia (<1% O2) (C), FIH is also inhibited and HIF is able to become fully active by the recruitment of co-activators such as p300.
Biochemical characterisation revealed that PHDs have low affinity for molecular oxygen. Low affinity for molecular oxygen signifies that when oxygen availability is reduced, these enzymes are quickly inhibited, leading to HIF stabilisation and activation of target genes. PHD inhibition in hypoxia has resulted in them being termed molecular oxygen sensors in the cell [12]. Additionally, many more 2-OGDs have been identified, most of which act independently of HIF but play a role in coordinating the cellular response to hypoxia. These include Factor Inhibiting HIF1 (FIH), Jumonji C (JmjC)-domain containing demethylases (JmjC demethylases) (which demethylate both histones and non-histone proteins), Ten-Eleven Translocation (TET) enzymes (mediators of DNA-demethylation), and RNA-demethylases (reported in vitro for 2-ODGs are shown is Table 1 and Supplementary Table S1). Given the function of some of these enzymes, it is conceivable that hypoxia could influence all aspects of gene expression, from chromatin structure and epigenetics to RNA biology, translation and protein turnover (Figure 2). This perfectly equips the cell when faced with hypoxia. Under such conditions, the cell must make a coordinated effort to allow for restoration of oxygen homeostasis, while reducing energy expenditure if it is to survive.
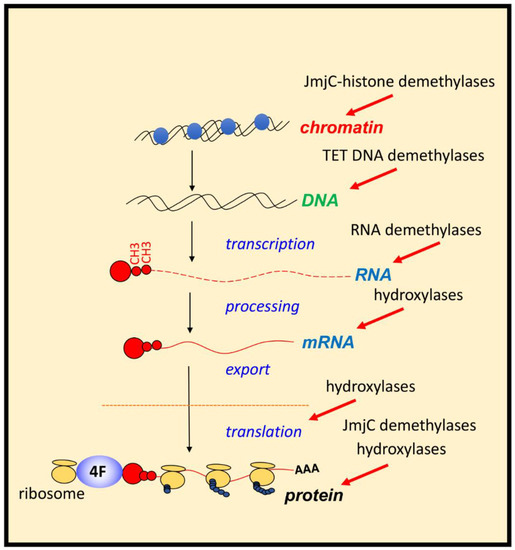
Figure 2. Control of gene expression by 2-ODGs in hypoxia. Hypoxia via the regulation of 2-OGDs has the potential of controlling all aspects of gene expression and protein function. Action of JmjC-histone demethylases and TETs (mediators of DNA demethylation) will impact on chromatin and DNA regulation. RNA demethylases, and several hydroxylases acts on mRNA processing, and fate. Hydroxylases also control rates of translation and ribosome activity, while JmjC-demethylases and hydroxylases can control protein function directly or indirectly by controlling other PTMs.
2. Effects of Hypoxia on Gene Transcription
It is now appreciated that the cellular and organism response to hypoxia involves profound changes to gene expression, with vast changes in gene transcription being detected in all systems studied.
Hypoxia-Induced Changes to Transcription Are Largely Mediated by HIF
As mentioned earlier HIFs are the master regulators of gene transcriptional changes in hypoxia and this has been extensively reviewed previously[2][11][13]. The discovery of hypoxia responsive genes and HIF targets has been driven greatly by transcript profiling and genome occupancy technologies, including microarrays, RNA-seq and ChIP-seq (reviewed in [14][15][16]). These analyses of publicly available transcriptional datasets[17][18] have shed light on cell type differences in hypoxia responsive genes and HIF targets on a genome wide scale, as well as identifying hypoxic signatures conserved across multiple cell types. Why different cell types have different transcriptional responses to hypoxia and HIF target genes is an important question for the field, with chromatin structure organisation thought to be a major factor in conferring specificity. Further to HIF isoform expression and activity, evidence points towards pre-established chromatin accessibility and local chromatin environment, including RNA pol II availability, pre-existing promoter enhancer interactions at HREs, and HRE DNA methylation status, as cell-type specificity determinants of hypoxia transcriptional responses (reviewed in [14][15][16][19]).
Whilst most HIF binding sites are at proximal promoters, binding to distal intergenic regions also occurs and HIF can regulate transcription of long genomic intervals, interacting at promoter-enhancer loops[20][21][22]. Although there is no doubt HIF is the main transcription factor controlling hypoxia-induced transcriptional changes, there is involvement of other transcription factors, including Nuclear Factor-κB (NF-κB), Tumor Protein p53 (p53), MYC Proto-Oncogene (MYC) and Activator Protein 1 (AP1), which function in the regulation of the hypoxia response via HIF-dependent and independent pathways (reviewed in[23]). In addition, HIF mostly acts as an activator of transcription, and thus most of the observed hypoxia-induced gene silencing is either independent of HIF, or via indirect mechanisms, including through the actions of chromatin remodeller complexes, co-repressor complexes or induction of other transcription factors (reviewed in[24]). HIF induced genes are involved in a variety of different pathways involved in restoration of oxygen homeostasis[2][11][13]. Importantly, many 2-OGDs, including PHD2[25] and PHD3[26], several JmjC demethylases (reviewed in[27]), and TET1 [28] and TET3 [28][29], are HIF target genes that have been shown to be transcriptionally upregulated in response to hypoxia.
Although we now have access to a vast number of transcriptomic studies of cells in hypoxia, very few studies have investigated the proteomic changes under such an important stress, using techniques ranging from the “old-fashioned” two-dimensional electrophoresis (2-DE) coupled with MALDI-TOF-TOF-MS [30][31][32], to the more accurate and robust quantitative multiplexed proteomics workflow[33][34][35][36], which usually combines isobaric labelling, two-dimensional liquid chromatography (2D-LC) and high resolution MS. The few studies available have shown that hypoxia exposure of different cells and tissues possesses broad effects at the whole proteome level, including changes to the protein expression of annexin family[30], glycolytic and antioxidant enzymes[31][32], transcription factors[37], heat shock proteins, S100 family proteins[33], and also other proteins involved in the TCA-cycle[38] , metabolism[39] and immune response [34]. A proteomic study has revealed novel non-HIF hypoxia regulators, including the chromatin organizer protein Heterochromatin Protein 1 Binding Protein 3 (HP1BP3), which mediates chromatin condensation [35] . The use of multi-omics techniques (transcriptomics, proteomics and metabolomics) and analysis using integrated bioinformatics identified consistent changes to proteins and metabolites in heart tissues under antenatal hypoxia. These proteins and metabolites are involved in energy metabolism, oxidative stress and inflammation-related pathways, required for the reprogramming of the mitochondrion[36].
3. Chromatin Regulation in Hypoxia
Central to the hypoxia response is the activation of a dynamic transcriptional programme. HIF transcription factors are the primary mediators of hypoxia induced gene transcriptional changes (reviewed in[11]). Further to HIF stabilisation and activation under low oxygen tensions, the chromatin landscape also plays a complex role in coordinating hypoxia inducible changes to gene transcription. Most aspects of chromatin regulation are altered in response to low oxygen, including histone methylation and acetylation, DNA methylation, actions of chromatin remodeller complexes and non coding RNAs, histone eviction and incorporation of histone variants, and chromatin accessibility (reviewed in [14][15][13]). However, this is still a vastly unexplored aspect of the hypoxia response. As mentioned above, in addition to PHDs, TETs (mediators of DNA demethylation), and JmjC demethylases (histone and non-histone protein demethylases), are also 2-OGDs. Recent studies demonstrate the potential of TETs and JmjC demethylases to function as molecular oxygen sensors, directly linking oxygen sensing to transcriptional control via epigenetics in cells. Below we summarise DNA and histone methylation changes in hypoxia, with a focus on oxygen sensing mechanisms via TETs and JmjC demethylases. (Figure 3).
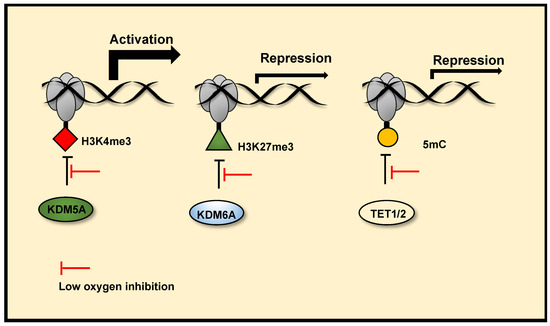
Figure 3. Chromatin oxygen sensing via JmjC histone demethylases and TETs. JmjC histone demethylases and TETs (mediators DNA demethylation) are 2-OGDs. Reduced activity of these enzymes in hypoxia, due to their oxygen sensitivity, can alter the chromatin landscape and mediate hypoxia induced transcription changes. Reduced activity of KDM5A in hypoxia increases H3K4me3 at the promoters of a subset hypoxia induced genes, facilitating their transcriptional activation. Reduced activity of KDM6A in hypoxia increases H3K27me3 at the promoters of a subset of hypoxia-repressed genes and represses their transcription. Reduced TET1/2 activity in hypoxia also represses gene transcription via DNA hypermethylation at gene promoters.
3.1. JmjC Demethylases and Chromatin Regulation in Hypoxia
Histone methylation is a recognised mechanism controlling chromatin structure and is associated with regulation of gene transcription, with some marks clearly leading to open chromatin, while others are firmly associated with closed conformation [40]. The family of enzymes predominantly responsible for histone demethylation are JmjC demethylases, which are part of the JmjC-domain containing group of 2-OGDs that includes demethylases and hydroxylases (JmjC 2-ODGs)). In vitro studies demonstrate the varied oxygen sensitivities of these enzymes, some are potentially direct molecular oxygen sensors (Table 1 and Supplementary Table S1). Oxygen affinities, oxygen availability and protein expression levels will likely dictate JmjC demethylase activities in hypoxia. Several groups have reported increases in total levels of histone methylation modifications in response to hypoxia across a range of human and mouse cell types and human tumours using immunoblotting, immunohistochemistry, immunofluorescence and quantitative proteomics ([41], reviewed in[15]). ChIP-sequencing approaches have revealed site-specific, hypoxia-induced changes in Histone (H)3 Lysine (K)4 trimethylation (me3)[42][43], H3K36me3[42] and H3K27me3 [44][43], which correlate with changes in gene expression. There is now evidence, through the use of in vitro and in cell histone demethylation assays, in coordination with mutagenesis analysis, gene expression analysis and histone methylation analysis, that Lysine Demethylase (KDM) 6A (KDM6A) [44] and potentially KDM5A[42], which demethylate H3K27me3 and H3K4me3 respectively, are inhibited by reduced oxygen levels in hypoxia. These result in increased histone methylation modifications, which coordinate hypoxia inducible gene transcriptional changes (Figure 3) and hypoxia induced cellular responses. Specifically, KDM6A inhibition in hypoxia triggers hypermethylation of H3K27me3 at a subset of hypoxia repressed gene promoters, reducing their expression. Conversely, potential KDM5A inhibition in hypoxia, triggers H3K4me3 hypermethylation at a subset of hypoxia inducible gene promoters, this precedes increases in their expression in hypoxia and is required for their full transcriptional activation in hypoxia. In cell and in vitro H3K9me3 demethylation assays have also revealed that the demethylase activity of KDM4A is highly sensitive to oxygen concentrations over physiologically relevant ranges [45]. Although there are a range of reported in vitro oxygen affinities, (Supplementary Table S1) the data from cells show that KDM4A activity is sensitive to oxygen levels. There is evidence that reduced histone demethylase activity of KDM4A in severe hypoxia (<0.5% oxygen), along with KDM4C, triggers increased expression of a subset of androgen receptor target genes[46]. Thus, KDM4A can also be classed as an oxygen sensor. Interestingly, KDM4A has been shown to positively regulate HIF-1α levels via H3K9me3 demethylation at the HIF1A gene locus, this effect is observed in mild hypoxia (2% oxygen), but impaired at severe hypoxia (<0.1% oxygen) [47]. This may provide a mechanism of maintaining HIF-1α levels in conditions of mild hypoxia specifically, where there is still residual PHD activity. Future work should investigate if the oxygen sensitive H3K9me3 demethylase activity of KDM4A is linked to control of gene expression and chromatin regulation in hypoxia. Importantly, some JmjC demethylases remain active at low oxygen concentrations and function in hypoxia through their histone demethylase activity. KDM4C [48] and KDM3A [49][50] display HIF coactivator activity in hypoxia via demethylation of H3K9 at HIF target gene promoters, facilitating transcriptional activation at these genes. The co-activator role of KDM3A in hypoxia, via retained histone demethylation activity appears to be context dependent, as other groups have reported reduced activity KDM3A in hypoxia[51]. Furthermore, many JmjC histone demethylases are HIF target genes that are upregulated in hypoxia (reviewed in [26]), this is thought in part to be a compensatory mechanism to counteract reduced demethylase activity, acting as a hypoxia feedback loop similar to what is seen with transcriptional upregulation of PHD2/3 by HIF. Thus, there is complex crosstalk between histone methylation, gene expression and hypoxia, mediated in part by JmjC demethylases. However, further characterisation of the oxygen sensitives of JmjC demethylases is needed.
3.2. TET Mediated DNA Demethylation Functions in Hypoxia
TETs, of which there are three variants—Ten Eleven Ten translocation Methylcytosine Dioxygenase 1 (TET1), TET2 and TET3—are 2-ODGs that function as hydroxylases, mediating mammalian DNA demethylation through catalysing the oxidation of 5-methylcytosine (5mc) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). These TET oxidised derivatives of 5mc can be demethylated by mechanisms of active and passive demethylation (reviewed in [52]). DNA methylation can repress gene transcription, consequently tumour hypoxia results in aberrant DNA methylation profiles promoting tumour suppressor gene silencing (hypermethylation)[53] and oncogene activation (hypomethylation)[54]. Recently, using in vitro biochemical binding assays, in vivo studies on HIF binding and DNA methylation status in human cancer cell lines, and in silico structural modelling, D’Anna and colleagues find that DNA methylation at HREs impairs HIF binding, and HRE DNA methylation status is a key factor in determining cell type specific transcriptional responses to hypoxia [55]. There is heterogeneity regarding the effects of hypoxia, both in cell models and in tumours, on global DNA methylation levels and TET activity (reviewed in [13][56]). As such, whether TETs are impaired or functionally active in hypoxia, and the consequences this has for gene transcription appear highly context dependent. Researchers have shown TET activity in hypoxia, and HIF-dependent TET upregulation and coactivator functions have been demonstrated at hypoxia-inducible genes (reviewed in [13][56]). TET activity in low oxygen environments is supported by in vitro oxygen affinities of TET1 and TET2 (Table 1 and Supplementary Table S1), as well as the known roles of TETs in the bone marrow and during development where oxygen tensions are low [57][58]. Conversely, Thienpont et al. showed that severe hypoxia (0.5% oxygen) in human and murine cells and tumour hypoxia in multiple human tumours causes DNA hypermethylation at gene promoters correlating with gene silencing at a subset of hypoxia repressed genes and gene silencing linked to hypoxia associated tumour progression. DNA hypermethylation was attributed to oxygen dependent reduction in TET1 and TET2 activity in hypoxia, with a 50% reduction in activity observed at 0.3% oxygen for TET1 and 0.5% for TET2 in vitro. Thus, TET1 and TET2 may be characterised as tumour oxygen sensors, and depending on the context of oxygen deprivation, may remain active in hypoxia environments or display inhibition. However, more work is needed to establish the oxygen dependence of TET activity in cells and in vivo and the physiological contexts in which TETs can sense changes in oxygen availability, as well as the consequences this has for DNA methylation, gene transcription and cellular responses. Indeed, the seemingly contradictory roles for TETs in hypoxia from studies to date may be dependent on the different cell models used and timing/severity of hypoxic stimulus.
While there is growing evidence for a dynamic role of chromatin/epigenetics in sensing and responding to hypoxia to facilitate transcriptional changes, via HIF-dependent and -independent mechanisms, efforts to elucidate molecular mechanisms underpinning such changes and the extent to which chromatin/epigenetic changes are required for coordinating hypoxia/HIF transcriptional effects are ongoing. The discoveries of oxygen sensing by TETs and JmjC demethylases provide an exciting link between oxygen availability and chromatin regulation, and future work on oxygen sensing by chromatin will be essential for better understanding hypoxia driven processes.
4. Effects of Hypoxia on Translation, focusing on roles of 2-OGDs
Protein levels and function are key aspects to achieve the appropriate hypoxia response. Although transcription is important, mechanisms controlling protein levels and function supersede any changes in transcriptional output. In hypoxia, mechanisms exist that control translation, but also post-translation aspects of protein function.
In addition to the regulation of gene transcription in hypoxia, gene expression is also controlled through regulation of translation (Figure 4). The cellular response to hypoxia includes a reduction in the energy expenditure of the cell due to limited ATP production through oxidative phosphorylation. This adaptation results in a reversible global decrease in energy-expensive protein synthesis (reviewed in [2]2]). This inhibition of translation is a highly regulated response to low oxygen levels preceding ATP depletion (reviewed in[2]).
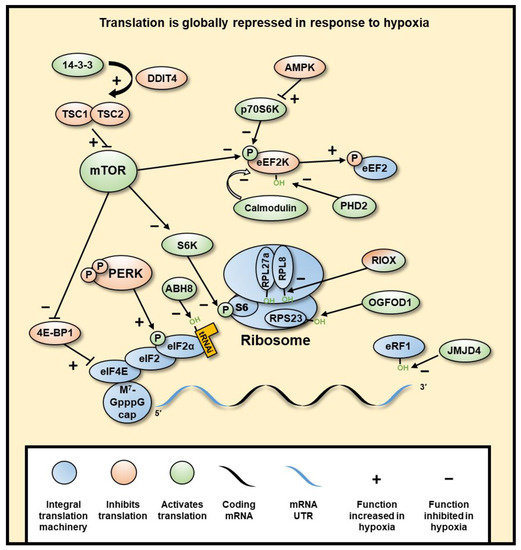
Figure 4. Hypoxia induces a global inhibition of protein translation. Global translation is mostly inhibited at initiation through mTOR inhibition of eIF4 and PERK inhibition of eIF2α, subsequently decreasing cap-dependent translation of mRNA. mTOR is inhibited through hypoxia-induced DDIT4-dependent release of TSC2 from 14-3-3 binding proteins, resulting in the inhibition of mTOR by TSC1/2 dimer. Elongation is also regulated through mTOR, as well as AMPK through its inhibition of RPS6KB1, wherein hypoxia eEF2K is not inhibited, which allows its phosphorylation and inhibition of eEF2. PHD2 can also hydroxylate eEF2K, which in normoxia causes its disassociation from calmodulin, decreasing its autophosphorylation. Termination is regulated by JMJD4-mediated hydroxylation of eRF1, which is required for termination. Selective translation of genes in hypoxia is regulated by UTR sequences, such as IRES and uORFs, which allow increased translation specifically in hypoxia. RNA binding proteins can bind to various parts of the mRNA and result in different regulatory outcomes. The ribosome can also be hydroxylated by RIOX1 and RIOX2, though it is not yet clear what role these modifications have.
Global inhibition of protein expression is largely regulated at the point of translation initiation through two pathways. Firstly, Mechanistic Target of Rapamycin Kinase (mTOR) is inhibited by DNA Damage Inducible Transcript 4 (DDIT4) (a HIF target gene). DDIT4-dependent release of TSC Complex Subunit 2 (TSC2) from 14-3-3 binding proteins leads to mTOR inhibition. This allows the formation of an active TSC1-TSC2 dimer that inhibits the phosphorylation of Ribosomal Protein S6 Kinase B1 (RPS6KB1) by the mTOR protein complex, which in turn inhibits the phosphorylation of Ribosomal Protein S6 (RPS6), part of the 40S ribosomal subunit required for initiation[59]. mTOR inhibition also causes hypophosphorylation of Eukaryotic Translation Initiation Factor (eIF) 4E Binding Protein 1 (eIF4EBP1), allowing the sequestering of eIF4E and decrease of 5’cap-dependent initiation[60][61]. Secondly, PKR-like Endoplasmic Reticulum Kinase (PERK) (gene name EIF2AK3) is phosphorylated and activated, which subsequently phosphorylates eIF2α at S51, causing effective inactivation[62][63]. Phospho-eIF2α prevents binding with eIF2B for exchange of GDP for GTP, therefore remaining in an inactive state and preventing subsequent rounds of translation from the mRNA [64]. eIF2α normally recruits the initiator aminoacylated tRNA to the 40S ribosome, thus limiting global initiation of translation. This second mechanism is independent of HIF and as of yet has not been linked to any 2-OGD.
Inhibition of translation is also regulated at the stage of polypeptide elongation. Elongation is inhibited by phosphorylation of Eukaryotic Elongation Factor 2 (eEF2) at T56 by eEF2 Kinase (eEF2K) [65]. This process has been shown to be dependent on mTOR and 5′-AMP-activated protein kinase catalytic subunit alpha-1 (AMPK) (gene name PRKAA1) [66]. Interestingly, eEF2 kinase (eEF2K) is also regulated by hydroxylation by PHD2 at P98 in an oxygen-dependent manner [67]. In hypoxia, when PHD2 inhibited, eEF2K activity is induced.
In addition to PHD2 dependent hydroxylation, there are several other hydroxylation reactions involved in the regulation of translation, catalysed by other 2-OGDs. Hydroxylation is important for the biosynthesis of tRNAPhe with position 37 requiring a hypermodified nucleoside Wybutosine (yW) that can be hydroxylated to form hydroxywybutosine (OHyW) by the JmjC hydroxylase, TRNA-YW Synthesizing Protein 5 (TYW5), which maintains translational fidelity. It is currently unknown whether TYW5 is responsive to the level of oxygen, but its transcription is decreased in hypoxia, [68] linking hypoxia to a decreased accuracy of translation, which globally decreases the successful translation of proteins[69].
The rate and accuracy of translation are positively regulated by hydroxylation of the central translation machinery. JmjC hydroxylases Ribosomal Oxygenase 2 (RIOX2) and RIOX1 hydroxylate histidyl residues in ribosomal proteins, with RIOX2 and RIOX1 hydroxylating Ribosomal Protein L (RPL) 27a (RPL27A) and (RPL8), respectively. The hydroxylation occurs at residues close to the peptidyl transfer centre, thereby increasing translation efficiency[70]. RIOX1 and RIOX2 transcription is reduced in hypoxia [68][70]. Furthermore, RPL8 hydroxylation is also reduced in hypoxia [70]. However, it is not yet known whether these enzymes are inhibited by low oxygen levels, or lower hydroxylation is solely due to lower transcription. Additionally, hydroxylation of 40S Ribosomal Protein S23 (RPS23) by the 2-OGD, 2-Oxoglutarate and Iron Dependent Oxygenase Domain Containing 1 (OGFOD1), is required for efficient translation [71]. OGFOD1 transcription is also decreased in hypoxia, but the enzyme remains mostly active, even in acute hypoxia[72], suggesting this mechanism is not through direct 2-OGD oxygen sensing.
Efficient decoding of the mRNA during translation requires the JmjC hydroxylase, AlkB Homolog 8, TRNA Methyltransferase, ALKBH8, which hydroxylates tRNA at the wobble position [73][74]. This 2-OGD has yet to be linked to hypoxia, though it would be interesting to investigate its oxygen sensitivity. Finally, lysyl hydroxylation of eukaryotic release factor 1 (eRF1) by the JmjC hydroxylase Jumonji Domain Containing 4 (JMJD4) is required for proper termination of translation [75], although its activity is not significantly inhibited in hypoxia.
5. Other Potential Roles of 2-OGDs in the Hypoxia Response
Further to known and potential oxygen sensing roles of JmjC 2-ODGs (JmjC demethylases and hydroxylases) in the regulation of chromatin and translation discussed earlier, there are other functions of JmjC 2-ODGs that may influence the hypoxia response (Figure 5). One of the most prominent of such enzymes is the JmjC 2-OGD, JMJD6, Arginine Demethylase and Lysine Hydroxylase, which has unique activities as both an arginine demethylase (acting on mono- and symmetric/asymmetric di-methylated arginines) and lysine hydroxylase[76][77]. JMJD6 expression is increased in hypoxic conditions in the placenta and can downregulate HIF-1α[78], though it has been found to operate in diverse pathways. JMJD6 can promote the formation of stress granules through demethylation of Arg-435, Arg-447 and Arg-460 of G3BP Stress Granule Assembly Fact 1 G3BP1, and subsequent de-repression of the protein, resulting in the cytoplasmic sequestering of stalled mRNA-ribosome complexes to reversibly prevent mRNA degradation[79][80]. This would allow a fast re-start of protein synthesis when oxygen homeostasis is restored. JMJD6 also regulates mRNA splicing through hydroxylating the splicing regulatory (SR) proteins LUC7 Like 2, Pre-MRNA Splicing Factor (LUC7L2) on Lys-269, U2 Small Nuclear RNA Auxiliary Factor 65 (U2AF65) on Lys-15 and Lys-276 [81], and multiple fragments of Serine and Arginine Rich Splicing Factor 11 (SRSF11) between residue 271 to 372 [76]. The SR proteins are involved in exon definition and alternative splicing, with SRSF11 hydroxylation resulting in skipping of most 5’ exon, and hydroxylation of U2AF65 possibly enacting pre-mRNA looping in order to present to the splicing machinery different cis splice enhancer or silencer sequences[76]. However, this only occurs for selected mRNAs and is not a global effect[82]. Nevertheless, this mechanism would allow selection of alternate splice variants as a response to hypoxia. JMJD6 can also interact with both Bromodomain Containing 4 (BRD4) and the positive Transcription Factor Elongation Factor b (P-TEFb) complex[83], eventually resulting in the release of paused DNA polymerase II and resumption of mRNA synthesis at specifically regulated genes . This implies that hypoxia could use this mechanism to stall transcription of genes that are not required for the stress response to hypoxia and would allow a re-start of gene expression when oxygen levels are restored. JMJD6 hydroxylates p53 on Lys382, repressing its transcriptional activity [84]. JMJD6 also auto-hydroxylates itself on the N-terminus and this is required for homo-oligomerisation[85]. The function of the homomultimer of JMJD6 is unknown, but speculated to play a role as a scaffolding protein in the nucleus [86].
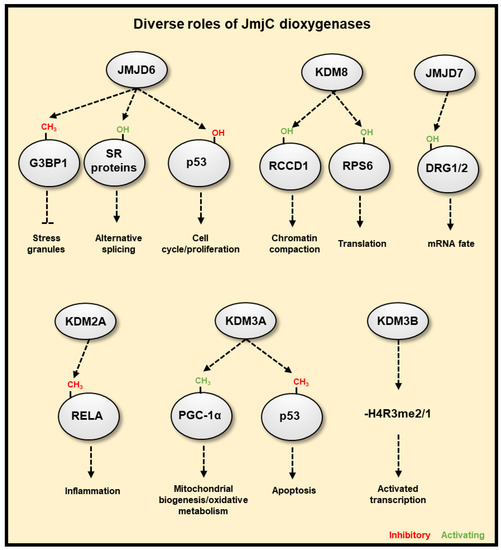
Figure 5. JmjC-domain containing 2OG dioxygenases have diverse cellular functions. JMJD6 is able to promote the formation of stress granules, which reversibly pauses transcript translation through demethylation of G3BP1. JMJD6 can hydroxylate splicing regulatory (SR) proteins, resulting in differential splicing or exon choice, such as skipping the first exon with the hydroxylation of SRSF11. JMJD6 can also hydroxylate p53 repressing its activity. KDM8 is an arginine hydroxylase that can target RCCD1 for chromatin condensation, and RPS6, which affects translation. JMJD7 is a lysyl hydroxylase that can target DRG1/2, affecting their regulation of RNA. KDM2A represses NF-κB activity via demethylation of RELA. KDM3A can demethylate both PGC-1α, promoting mitochondrial biogenesis, and p53, inhibiting its proapoptotic activity. KDM3B is able to demethylate mono- or di-methylated arginine residues, which functions to activate gene transcription.
Another JmjC hydroxylase, KDM8, can hydroxylate arginine residues in both RCC1 Domain Containing 1 (RCCD1) and RPS6[87]. KDM8 also has amine- and endo-peptidase activity with specificity towards histone H3 monomethyl-lysine residues, especially of the histone variant H3F3A[88], and arginine methylated histone tails[89][90]. It had previously been reported that KDM8 can demethylate H3K36me2[91], though considering its arginine demethylation is achieved through trimming of the histone tail, this may be the mechanism of observed decreases in lysine methylation. Although not necessarily dependent on its hydroxylation activity, KDM8 is required for cell proliferation and chromosomal stability [92], and can negatively regulate p53, affecting gene expression and control cell cycle and proliferation[93][94]. Recently, a biochemical function has been assigned to JMJD7 as a lysyl hydroxylase, which targets Developmentally Regulated GTP Binding Protein 1 (DRG1) and DRG2 that are part of the Translation Factor (TRAFAC) family of GTPases, and could affect their binding with messenger, or ribosomal RNA, though this requires further investigation [95]. Similar to KDM8, JMJD7 also possesses endo- and amine-peptidase activity, preferentially cleaving mono- and di-methylated arginine residues of H2, H3, and H4[88][89].
Several JmjC demethylases have been found to have additional, non-histone substrates. For example, KDM2A represses NF-κB activity via demethylation of RELA, providing a possible link to hypoxia and inflammation crosstalk. [96]. KDM3A was recently found to function as a molecular oxygen sensor, with its demethylation of K224me1 on peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) (which facilitates its activity) impaired under low oxygen conditions [51]. PGC-1α is a transcriptional coactivator promoting mitochondrial biogenesis and oxidative metabolism, the aforementioned work neatly links mitochondrial function to the hypoxia response via the oxygen sensing function of KDM3A controlling PGC-1α activity, further highlighting the importance of JmjC 2-OGDs in the hypoxia response. KDM3A can also demethylate K372me1 of p53, which decreases its association with chromatin and represses its proapoptotic activity [97]. KDM3B, in addition to its substrate H3K9me2, can also demethylate mono- or di-methylated arginine residues, H4R3me2[98]. It is more than likely that other JmjC demethylases interact and directly demethylate additional transcription factors which may coordinate transcriptional responses to hypoxia. However, unbiased analysis is required to fully assess this aspect of hypoxia-induced gene regulation. The KDM4 family of demethylases appears to have higher activity against trimethylated non-histone targets than its canonical substrates, H3K9me2 and H3K36me3[99]. These substrates include widely interspaced zinc finger motifs protein (WIZ), chromodomain Y-like protein (CDYL1), Cockayne syndrome group B protein (CSB, four peptides) and Histone-lysine N-methyltransferase (EHMT2). These substrates of KDM4 family are all members of transcription repression complexes, suggesting an additional role of transcriptional regulation by these demethylases. Using in vitro assays, KDM3A, KDM4E, KDM5C, and KDM6B have been shown to possess arginine demethylase activity, in addition to their lysine demethylase activity on peptide substrates[100]. Non-specific hydroxylase activities are not unusual for JmjC enzymes, so any such reported activity should demonstrate specificity and biological relevance, though it is possible that it will emerge that many lysine demethylases also possess relevant arginine demethylase activity.
Finally, a potentially new and exciting mechanism involves an epigenetic mark in RNA, methylation of 6 adenosine (m6A). mRNA m6A modification at the 5’UTR can recruit m6A binding proteins such as YTH N6-Methyladenosine RNA Binding Protein 2 (YTHDF2) in heat shock stress, which upregulates translation through binding eIF3 and the 40S subunit[101], and such a mechanism may prove true in other stress responses, such as hypoxia. The RNA demethylating enzymes FTO Alpha-Ketoglutarate Dependent Dioxygenase (FTO) and AlkB Homolog 5, RNA Demethylase (ALKBH5), are 2-OGDs, and although their ability to sense oxygen has yet to be investigated, their inhibition in hypoxia could result in an increase in global RNA methylation, which could regulate translation and RNA fate in hypoxia.
6. Relevance to Human Biology and Health
Although we currently do not know the importance of all of the 2-OGDs present in the genome, several of the key players in the hypoxia response have important functions and relevance to human biology and health. This is exemplified by the phenotypes observed in null mice, or by the presence of disease associated mutations in humans.
6.1. PHD/HIF/VHL Axis
Given the cellular functions mentioned above, it is no surprise that genetic mutations of most dioxygenases and HIFs have been implicated with human diseases. Mutations in HIF-2α and PHD2 have been found in patients with vascular pathologies, such as erythrocytosis, polycythemia, and pheochromocytoma (Table 2). As HIF mediates hypoxia adaptation responses, including the regulation of erythropoiesis and vasculogenesis, it is not surprising that mutations within the PHD/HIF/VHL axis are associated with vascular pathologies. The crucial role of HIFs in vascular pathologies is strongly demonstrated by genetic studies of mice, as highlighted in Supplementary Table S2. Knockout mice of HIF-1α or HIF-2α are embryonic lethal with vascular defects (Table 2, Supplementary Table S2), whereas the deletion of PHD2 that activates HIF signalling results in embryonic lethality in mice due to placental and heart defects (Table 2, Supplementary Table S2). VHL mutations also result in highly vascularized tumours, including pheochromocytomas, renal cell cancer carcinoma, retinal and central nervous system hemangioblastomas (Table 2). Hundreds of VHL mutations have been identified in VHL syndrome patients (listed in the Human Gene Mutation Database [102]). The homozygous VHL mutation R200W, which prevents efficient HIF-α degradation in normoxia, is found in all individuals with Chuvash Polycythemia (CP) [103]. CP is characterized by congenital erythrocytosis, and patients have been associated with pulmonary hypertension, thrombosis, vertebral hemangiomas, cerebral vascular events and other vascular abnormalities[104][105], displaying the role of VHL in HIF-dependent regulation of vasculogenesis and erythropoiesis.
6.2. 2-OGDs—Hydroxylases
Similar to PHDs, prolyl-4-hydroxylases P4HA1 and P4HA2 are hypoxia-inducible, but P4HA1/2 prolyl hydroxylation is required for different processes, that is collagen fiber formation. Consistent with their roles in collagen synthesis, P4HA1 and P4HA2 mutations are found in patients with collagen-related extracellular matrix disorders (Table 2; Supplementary Table S2). Furthermore, homozygous deletion of P4HA1 is embryonic lethal with base membrane rupture due to defective collagen IV assembly (Table 2). PAHX is another hydroxylase, but of phytanoyl-CoA; essential for breaking down phytanic fatty acid. Mutations in PAHX is well associated with Refsum disease, a rare inherited neurological disorder caused by neurotoxic phytanic acid as these mutations result in an enzymatically inactive protein, thus leading to phytanic acid accumulation.
The roles of TET1–3 in development are demonstrated in knockout mouse models (reviewed in[106]). TET1-null mice present several defects but these depend on the mode of genetic deletion. TET3 deletion results in neonatal lethality, highlighting TET3 role in development. TET3 mutations have been found in patients with intellectual disability and/or delayed global development (Table 2; Supplementary Table S2). Although somatic alterations of TET2 have been found in several cancers, these mutations are majorly associated with myelodysplastic syndromes (Table 2; Supplementary Table S2). In addition to the listed mutations in Supplementary Table S2, a study reported TET2 somatic mutations in 46 patients with myelodysplastic syndromes, myeloproliferative disorder, secondary acute myeloid leukemia, or chronic myelomonocytic leukemia [107]. Most of these mutations are predicted to lead to partial or total loss of function due to protein truncation.
6.3. 2-OGDs—JmjC Demethylases
Many of the JmjC demethylase genes have been associated with human diseases. In particular, several of them are mutated in patients with neurodevelopmental disorders, midline defects and cancers (Table 2; Supplementary Table S2). Although KDM3A is found to be mutated in infertile males[108], its role in infertility is not clear. Mutations in KDM3B are frequently implicated with intellectual disability, but also found in cancers including myeloid leukemias (Table 2). Similarly, JMJD1C mutations have been identified in individuals with autism spectrum disorder and intellectual disability. JMJD1C is also associated with congenital heart disease manifestation in 22q11.2 deletion syndrome patients. Amongst KDM4s, there are only two reports—single nucleotide substitutions of KDM4C in upper aerodigestive tract cancer and age of menarche. Mutations of KDM5B, KDM5C and KDM6B have been associated with neurodevelopment and a global developmental delay (Table 2; Supplementary Table S2). In particular, KDM5C is well recognised as an X-linked intellectual disability gene that is highly expressed in neural tissue. Mutations in PHD Finger Protein 8 (PHF8) are also associated with X-linked mental retardation and often accompanied by cleft lip/palate or autism. The phenotypes of KDM5C or PHF8 mutations in humans are reflected by the deletion of these genes in mice (Table 2). On the other hand, KDM6A mutations are frequently found in individuals with Kabuki syndrome (KS), a genetic disease with developmental delay and congenital anomalies, (Table 2) highlighting the role of KDM6A in development. In addition to mutations listed in Supplementary Table S2, others have reported gross deletions, gross duplications, or chromosomal rearrangement in patients with KS or KS-like clinical manifestations[109][110][111][112][113]. However, whether the phenotypes observed are due to loss of demethylase activity solely is currently unknown.
Oxygen regulation is pivotal for embryogenesis as most developmental processes during early embryo development occurs in a hypoxic environment[114]. Given the fact that 2-OGDs sense and respond to oxygen levels, it is not surprising that mutations affecting the activity of several of these enzymes are detrimental to embryo development, contributing to neonatal defects. The hypoxic environment is also a common feature of tumours due to aberrant vascularisation and an inadequate supply of blood [115]. Mutations or deletions of some of these 2-OGDs are implicated in cancers. The molecular mechanisms through which 2-OGDs promote cancers is not completely understood. However, altered metabolism in cancer has been linked to alteration of 2-OGDs function (reviewed in [116]). 2-OGDs can be inhibited by metabolites that are accumulated in tumours, including succinate, fumarate and D/L-2-hydroxyglutarate[115][117][116]. It is thus very likely that the phenotypes observed in deletions and mutations are the result of both alteration of the hypoxia response and changes in metabolism. Overall, the presence and connections of HIFs or dioxygenase mutations in human disorders and the knockout studies demonstrate the essential roles of these genes.
References
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythro-poietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454, doi:10.1128/mcb.12.12.5447.
- Rocha, S. Gene regulation under low oxygen: Holding your breath for transcription. Trends Biochem. Sci. 2007, 32, 389–397, doi:10.1016/j.tibs.2007.06.005.
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–269, doi:10.1152/ajpcell.00315.2015.
- Kliewe, F.; Engelhardt, M.; Aref, R.; Schuller, H.J. Promoter recruitment of corepressors Sin3 and Cyc8 by activator proteins of the yeast Saccharomyces cerevisiae. Curr. Genet. 2017, 63, 739–750, doi:10.1007/s00294-017-0677-8.
- D'Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-kappaB. Biomedicines 2017, 5, 21, doi:10.3390/biomedicines5020021.
- Biddlestone, J.; Bandarra, D.; Rocha, S. The role of hypoxia in inflammatory disease (review). Int. J. Mol. Med. 2015, 35, 859–869, doi:10.3892/ijmm.2015.2079.
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O'Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54, doi:10.1016/s0092-8674(01)00507-4.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468, doi:10.1126/science.1059817.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275, doi:10.1038/20459.
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427, doi:10.1038/35017054.
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402, doi:10.1016/j.molcel.2008.04.009.
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the ac-tivity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2007, 401, 227–234, doi:10.1042/BJ20061151.
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298, doi:10.1016/j.cmet.2017.10.005.
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020, 10.1111/febs.15374, doi:10.1111/febs.15374.
- Batie, M.; Rocha, S. Gene transcription and chromatin regulation in hypoxia. Biochem. Soc. Trans. 2020, 10.1042/BST20191106, doi:10.1042/BST20191106.
- Schodel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659, doi:10.1038/s41581-019-0182-z.
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; del Peso, L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010, 38, 2332–2345, doi:10.1093/nar/gkp1205.
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602, doi:10.1093/nar/gkp425.
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15, doi:10.3109/10409238.2013.838205.
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–217, doi:10.1182/blood-2010-10-314427.
- Platt, J.L.; Salama, R.; Smythies, J.; Choudhry, H.; Davies, J.O.; Hughes, J.R.; Ratcliffe, P.J.; Mole, D.R. Capture-C reveals preformed chromatin interactions between HIF-binding sites and distant promoters. EMBO Rep. 2016, 17, 1410–1421, doi:10.15252/embr.201642198.
- Orlando, I.M.C.; Lafleur, V.N.; Storti, F.; Spielmann, P.; Crowther, L.; Santambrogio, S.; Schodel, J.; Hoogewijs, D.; Mole, D.R.; Wenger, R.H. Distal and proximal hypoxia response elements cooperate to regulate organ-specific erythropoietin gene expression. Haematologica 2019, doi:10.3324/haematol.2019.236406.
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29, doi:10.1042/BJ20081055.
- Batie, M.; Del Peso, L.; Rocha, S. Hypoxia and Chromatin: A Focus on Transcriptional Repression Mechanisms. Biomedicines 2018, 6, 47, doi:10.3390/biomedicines6020047.
- Metzen, E.; Stiehl, D.P.; Doege, K.; Marxsen, J.H.; Hellwig-Burgel, T.; Jelkmann, W. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: Identification of a functional hypoxia-responsive element. Biochem. J. 2005, 387, 711–717, doi:10.1042/BJ20041736.
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landazuri, M.O.; Del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197, doi:10.1042/BJ20042121.
- Shmakova, A.; Batie, M.; Druker, J.; Rocha, S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem. J. 2014, 462, 385–395, doi:10.1042/BJ20140754.
- Wu, M.Z.; Chen, S.F.; Nieh, S.; Benner, C.; Ger, L.P.; Jan, C.I.; Ma, L.; Chen, C.H.; Hishida, T.; Chang, H.T.; et al. Hypoxia Drives Breast Tumor Malignancy through a TET-TNFalpha-p38-MAPK Signaling Axis. Cancer Res. 2015, 75, 3912–3924, doi:10.1158/0008-5472.CAN-14-3208.
- Cao, J.Z.; Liu, H.; Wickrema, A.; Godley, L.A. HIF-1 directly induces TET3 expression to enhance 5-hmC density and induce erythroid gene expression in hypoxia. Blood Adv. 2020, 4, 3053–3062, doi:10.1182/bloodadvances.2020001535.
- Hu, R.; Jin, H.; Zhou, S.; Yang, P.; Li, X. Proteomic analysis of hypoxia-induced responses in the syncytialization of human placental cell line BeWo. Placenta 2007, 28, 399–407, doi:10.1016/j.placenta.2006.07.005.
- Sharma, N.K.; Sethy, N.K.; Bhargava, K. Comparative proteome analysis reveals differential regulation of glycolytic and antioxidant enzymes in cortex and hippocampus exposed to short-term hypobaric hypoxia. J. Proteom. 2013, 79, 277–298, doi:10.1016/j.jprot.2012.12.020.
- Hoang, V.M.; Foulk, R.; Clauser, K.; Burlingame, A.; Gibson, B.W.; Fisher, S.J. Functional proteomics: Examining the effects of hypoxia on the cytotrophoblast protein repertoire. Biochemistry 2001, 40, 4077–4086, doi:10.1021/bi0023910.
- Li, Q.; Luo, T.; Lu, W.; Yi, X.; Zhao, Z.; Liu, J. Proteomic analysis of human periodontal ligament cells under hypoxia. Proteome Sci. 2019, 17, 3, doi:10.1186/s12953-019-0151-2.
- Dou, L.; Yan, Q.; Liang, P.; Zhou, P.; Zhang, Y.; Ji, P. iTRAQ-Based Proteomic Analysis Exploring the Influence of Hypoxia on the Proteome of Dental Pulp Stem Cells under 3D Culture. Proteomics 2018, 18, doi:10.1002/pmic.201700215.
- Dutta, B.; Ren, Y.; Hao, P.; Sim, K.H.; Cheow, E.; Adav, S.; Tam, J.P.; Sze, S.K. Profiling of the Chromatin-associated Proteome Identifies HP1BP3 as a Novel Regulator of Cell Cycle Progression. Mol. Cell. Proteom. 2014, 13, 2183–2197, doi:10.1074/mcp.M113.034975.
- Gao, Y.; Dasgupta, C.; Huang, L.; Song, R.; Zhang, Z.; Zhang, L. Multi-Omics Integration Reveals Short and Long-Term Effects of Gestational Hypoxia on the Heart Development. Cells 2019, 8, 608, doi:10.3390/cells8121608.
- Shakib, K.; Norman, J.T.; Fine, L.G.; Brown, L.R.; Godovac-Zimmermann, J. Proteomics profiling of nuclear proteins for kidney fibroblasts suggests hypoxia, meiosis, and cancer may meet in the nucleus. Proteomics 2005, 5, 2819–2838, doi:10.1002/pmic.200401108.
- Vodisch, M.; Scherlach, K.; Winkler, R.; Hertweck, C.; Braun, H.P.; Roth, M.; Haas, H.; Werner, E.R.; Brakhage, A.A.; Kniemeyer, O. Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin A biosynthesis gene cluster in response to hypoxia. J. Proteome Res. 2011, 10, 2508–2524, doi:10.1021/pr1012812.
- Janker, L.; Mayer, R.L.; Bileck, A.; Kreutz, D.; Mader, J.C.; Utpatel, K.; Heudobler, D.; Agis, H.; Gerner, C.; Slany, A. Metabolic, Anti-apoptotic and Immune Evasion Strategies of Primary Human Myeloma Cells Indicate Adaptations to Hypoxia. Mol. Cell. Proteom. 2019, 18, 936–953, doi:10.1074/mcp.RA119.001390.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705, doi:10.1016/j.cell.2007.02.005.
- Hsu, K.F.; Wilkins, S.E.; Hopkinson, R.J.; Sekirnik, R.; Flashman, E.; Kawamura, A.; McCullagh, J.S.O.; Walport, L.J.; Schofield, C.J. Hypoxia and hypoxia mimetics differentially modulate histone post-translational modifications. Epigenetics 2020, doi:10.1080/15592294.2020.1786305.
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226, doi:10.1126/science.aau5870.
- Prickaerts, P.; Adriaens, M.E.; Beucken, T.V.; Koch, E.; Dubois, L.; Dahlmans, V.E.; Gits, C.; Evelo, C.T.; Chan-Seng-Yue, M.; Wouters, B.G.; et al. Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenet. Chromatin 2016, 9, 46, doi:10.1186/s13072-016-0086-0.
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jen-nings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Sci-ence 2019, 363, 1217–1222, doi:10.1126/science.aaw1026.
- Hancock, R.L.; Masson, N.; Dunne, K.; Flashman, E.; Kawamura, A. The Activity of JmjC Histone Lysine Demethylase KDM4A is Highly Sensitive to Oxygen Concentrations. ACS Chem. Biol. 2017, 12, 1011–1019, doi:10.1021/acschembio.6b00958.
- Lee, H.Y.; Yang, E.G.; Park, H. Hypoxia enhances the expression of prostate-specific antigen by modifying the quantity and catalytic activity of Jumonji C domain-containing histone demethylases. Carcinogenesis 2013, 34, 2706–2715, doi:10.1093/carcin/bgt256.
- Dobrynin, G.; McAllister, T.E.; Leszczynska, K.B.; Ramachandran, S.; Krieg, A.J.; Kawamura, A.; Hammond, E.M. KDM4A regulates HIF-1 levels through H3K9me3. Sci. Rep. 2017, 7, 11094, doi:10.1038/s41598-017-11658-3.
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367-3376, doi:10.1073/pnas.1217394109.
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2010, 30, 344–353, doi:10.1128/MCB.00444-09.
- Wan, W.; Peng, K.; Li, M.; Qin, L.; Tong, Z.; Yan, J.; Shen, B.; Yu, C. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1alpha. Oncogene 2017, 36, 3868–3877, doi:10.1038/onc.2017.13.
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A Senses Oxygen Availability to Regulate PGC-1alpha-Mediated Mitochondrial Biogenesis. Mol. Cell. 2019, 76, 885–895 e887, doi:10.1016/j.molcel.2019.09.019.
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534, doi:10.1038/nrg.2017.33.
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D'Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquiere, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68, doi:10.1038/nature19081.
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007, 2, 119–125, doi:10.4161/epi.2.2.4613.
- D'Anna, F.; Van Dyck, L.; Xiong, J.; Zhao, H.; Berrens, R.V.; Qian, J.; Bieniasz-Krzywiec, P.; Chandra, V.; Schoonjans, L.; Matthews, J.; et al. DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 2020, 21, 182, doi:10.1186/s13059-020-02087-z.
- Ito, S.; D'Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133, doi:10.1038/nature09303.
- Camuzi, D.; de Amorim, I.S.S.; Ribeiro Pinto, L.F.; Oliveira Trivilin, L.; Mencalha, A.L.; Soares Lima, S.C. Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells 2019, 8, 300, doi:10.3390/cells8040300.
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935, doi:10.1126/science.1170116.
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904, doi:10.1101/gad.1256804.
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965, doi:10.1128/MCB.26.10.3955-3965.2006.
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745, doi:10.1016/j.cell.2009.01.042.
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416, doi:10.1128/mcb.22.21.7405-7416.2002.
- Noma, A.; Ishitani, R.; Kato, M.; Nagao, A.; Nureki, O.; Suzuki, T. Expanding role of the jumonji C domain as an RNA hydroxylase. J. Biol. Chem. 2010, 285, 34503–34507, doi:10.1074/jbc.M110.156398.
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell. Mol. Life Sci.2013, 70, 3493-3511, doi:10.1007/s00018-012-1252-6.
- Ryazanov, A.G.; Davydova, E.K. Mechanism of elongation factor 2 (EF-2) inactivation upon phosphorylation. Phosphorylated EF-2 is unable to catalyze translocation. FEBS Lett. 1989, 251, 187–190, doi:10.1016/0014-5793(89)81452-8.
- Browne, G.J.; Proud, C.G. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell. Biol. 2004, 24, 2986–2997, doi:10.1128/mcb.24.7.2986-2997.2004.
- Moore, C.E.; Mikolajek, H.; Regufe da Mota, S.; Wang, X.; Kenney, J.W.; Werner, J.M.; Proud, C.G. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell. Biol. 2015, 35, 1788–1804, doi:10.1128/MCB.01457-14.
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 2008, 416, 387–394, doi:10.1042/BJ20081238.
- Kato, M.; Araiso, Y.; Noma, A.; Nagao, A.; Suzuki, T.; Ishitani, R.; Nureki, O. Crystal structure of a novel JmjC-domain-containing protein, TYW5, involved in tRNA modification. Nucleic Acids Res. 2011, 39, 1576–1585, doi:10.1093/nar/gkq919.
- Ge, W.; Wolf, A.; Feng, T.; Ho, C.H.; Sekirnik, R.; Zayer, A.; Granatino, N.; Cockman, M.E.; Loenarz, C.; Loik, N.D.; et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat. Chem. Biol. 2012, 8, 960–962, doi:10.1038/nchembio.1093.
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.A.; Cockman, M.E.; Kessler, B.M.; Ratcliffe, P.J.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024, doi:10.1073/pnas.1311750111.
- Singleton, R.S.; Liu-Yi, P.; Formenti, F.; Ge, W.; Sekirnik, R.; Fischer, R.; Adam, J.; Pollard, P.J.; Wolf, A.; Thalhammer, A.; et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc. Natl. Acad. Sci. USA 2014, 111, 4031–4036, doi:10.1073/pnas.1314482111.
- Fu, D.; Brophy, J.A.; Chan, C.T.; Atmore, K.A.; Begley, U.; Paules, R.S.; Dedon, P.C.; Begley, T.J.; Samson, L.D. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol. Cell. Biol. 2010, 30, 2449–2459, doi:10.1128/MCB.01604-09.
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell. 2006, 21, 521–531, doi:10.1016/j.molcel.2006.01.010.
- Feng, T.; Yamamoto, A.; Wilkins, S.E.; Sokolova, E.; Yates, L.A.; Munzel, M.; Singh, P.; Hopkinson, R.J.; Fischer, R.; Cockman, M.E.; et al. Optimal translational termination requires C4 lysyl hydroxylation of eRF1. Mol. Cell. 2014, 53, 645–654, doi:10.1016/j.molcel.2013.12.028.
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447, doi:10.1126/science.1145801.
- Islam, M.S.; McDonough, M.A.; Chowdhury, R.; Gault, J.; Khan, A.; Pires, E.; Schofield, C.J. Biochemical and structural investigations clarify the substrate selectivity of the 2-oxoglutarate oxygenase JMJD6. J. Biol. Chem. 2019, 294, 11637–11652, doi:10.1074/jbc.RA119.008693.
- Alahari, S.; Post, M.; Caniggia, I. Jumonji Domain Containing Protein 6: A Novel Oxygen Sensor in the Human Placenta. Endocrinology 2015, 156, 3012–3025, doi:10.1210/en.2015-1262.
- Tsai, W.C.; Reineke, L.C.; Jain, A.; Jung, S.Y.; Lloyd, R.E. Histone arginine demethylase JMJD6 is linked to stress granule assembly through demethylation of the stress granule-nucleating protein G3BP1. J. Biol. Chem. 2017, 292, 18886–18896, doi:10.1074/jbc.M117.800706.
- Tsai, W.C.; Gayatri, S.; Reineke, L.C.; Sbardella, G.; Bedford, M.T.; Lloyd, R.E. Arginine Demethylation of G3BP1 Promotes Stress Granule Assembly. J. Biol. Chem. 2016, 291, 22671–22685, doi:10.1074/jbc.M116.739573.
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R., 3rd; et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93, doi:10.1126/science.1175865.
- Kwok, J.; O'Shea, M.; Hume, D.A.; Lengeling, A. Jmjd6, a JmjC Dioxygenase with Many Interaction Partners and Pleiotropic Functions. Front. Genet. 2017, 8, 32, doi:10.3389/fgene.2017.00032.
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595, doi:10.1016/j.cell.2013.10.056.
- Wang, F.; He, L.; Huangyang, P.; Liang, J.; Si, W.; Yan, R.; Han, X.; Liu, S.; Gui, B.; Li, W.; et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol. 2014, 12, e1001819, doi:10.1371/journal.pbio.1001819.
- Han, G.; Li, J.; Wang, Y.; Li, X.; Mao, H.; Liu, Y.; Chen, C.D. The hydroxylation activity of Jmjd6 is required for its homo-oligomerization. J. Cell. Biochem. 2012, 113, 1663–1670, doi:10.1002/jcb.24035.
- Tibrewal, N.; Liu, T.; Li, H.; Birge, R.B. Characterization of the biochemical and biophysical properties of the phosphatidylserine receptor (PS-R) gene product. Mol. Cell. Biochem. 2007, 304, 119–125, doi:10.1007/s11010-007-9492-8.
- Wilkins, S.E.; Islam, M.S.; Gannon, J.M.; Markolovic, S.; Hopkinson, R.J.; Ge, W.; Schofield, C.J.; Chowdhury, R. JMJD5 is a human arginyl C-3 hydroxylase. Nat. Commun. 2018, 9, 1180, doi:10.1038/s41467-018-03410-w.
- Shen, J.; Xiang, X.; Chen, L.; Wang, H.; Wu, L.; Sun, Y.; Ma, L.; Gu, X.; Liu, H.; Wang, L.; et al. JMJD5 cleaves monomethylated histone H3 N-tail under DNA damaging stress. EMBO Rep. 2017, 18, 2131–2143, doi:10.15252/embr.201743892.
- Liu, H.; Wang, C.; Lee, S.; Deng, Y.; Wither, M.; Oh, S.; Ning, F.; Dege, C.; Zhang, Q.; Liu, X.; et al. Clipping of arginine-methylated histone tails by JMJD5 and JMJD7. Proc. Natl. Acad. Sci. USA 2017, 114, E7717-E7726, doi:10.1073/pnas.1706831114.
- Liu, H.; Wang, C.; Lee, S.; Ning, F.; Wang, Y.; Zhang, Q.; Chen, Z.; Zang, J.; Nix, J.; Dai, S.; et al. Specific Recognition of Arginine Methylated Histone Tails by JMJD5 and JMJD7. Sci. Rep. 2018, 8, 3275, doi:10.1038/s41598-018-21432-8.
- Hsia, D.A.; Tepper, C.G.; Pochampalli, M.R.; Hsia, E.Y.; Izumiya, C.; Huerta, S.B.; Wright, M.E.; Chen, H.W.; Kung, H.J.; Izumiya, Y. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 9671–9676, doi:10.1073/pnas.1000401107.
- Marcon, E.; Ni, Z.; Pu, S.; Turinsky, A.L.; Trimble, S.S.; Olsen, J.B.; Silverman-Gavrila, R.; Silverman-Gavrila, L.; Phanse, S.; Guo, H.; et al. Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Rep. 2014, 8, 297–310, doi:10.1016/j.celrep.2014.05.050.
- Huang, X.; Zhang, S.; Qi, H.; Wang, Z.; Chen, H.W.; Shao, J.; Shen, J. JMJD5 interacts with p53 and negatively regulates p53 function in control of cell cycle and proliferation. Biochim. Biophys. Acta 2015, 1853, 2286–2295, doi:10.1016/j.bbamcr.2015.05.026.
- Ishimura, A.; Terashima, M.; Tange, S.; Suzuki, T. Jmjd5 functions as a regulator of p53 signaling during mouse embryogenesis. Cell Tissue Res. 2016, 363, 723–733, doi:10.1007/s00441-015-2276-7.
- Markolovic, S.; Zhuang, Q.; Wilkins, S.E.; Eaton, C.D.; Abboud, M.I.; Katz, M.J.; McNeil, H.E.; Lesniak, R.K.; Hall, C.; Struwe, W.B.; et al. The Jumonji-C oxygenase JMJD7 catalyzes (3S)-lysyl hydroxylation of TRAFAC GTPases. Nat. Chem. Biol. 2018, 14, 688–695, doi:10.1038/s41589-018-0071-y.
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51, doi:10.1073/pnas.0912493107.
- Ramadoss, S.; Guo, G.; Wang, C.Y. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 2017, 36, 47–59, doi:10.1038/onc.2016.174.
- Li, S.; Ali, S.; Duan, X.; Liu, S.; Du, J.; Liu, C.; Dai, H.; Zhou, M.; Zhou, L.; Yang, L.; et al. JMJD1B Demethylates H4R3me2s and H3K9me2 to Facilitate Gene Expression for Development of Hematopoietic Stem and Progenitor Cells. Cell Rep. 2018, 23, 389–403, doi:10.1016/j.celrep.2018.03.051.
- Li, S.; Ali, S.; Duan, X.; Liu, S.; Du, J.; Liu, C.; Dai, H.; Zhou, M.; Zhou, L.; Yang, L.; et al. JMJD1B Demethylates H4R3me2s and H3K9me2 to Facilitate Gene Expression for Development of Hematopoietic Stem and Progenitor Cells. Cell Rep. 2018, 23, 389–403, doi:10.1016/j.celrep.2018.03.051.
- Ponnaluri, V.K.; Vavilala, D.T.; Putty, S.; Gutheil, W.G.; Mukherji, M. Identification of non-histone substrates for JMJD2A-C histone demethylases. Biochem. Biophys. Res. Commun. 2009, 390, 280–284, doi:10.1016/j.bbrc.2009.09.107.
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974, doi:10.1038/ncomms11974.
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5' UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010, doi:10.1016/j.cell.2015.10.012.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9, doi:10.1007/s00439-013-1358-4.
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621, doi:10.1038/ng1019.
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932, doi:10.1182/blood-2003-07-2535.
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290, doi:10.1371/journal.pmed.0030290.
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750, doi:10.1101/gad.276568.115.
- Hojati, Z.; Soleimanpour, E.; Javadirad, S.M.; Nasr-Esfahani, M.H. Identification of Two Novel Mutations in KDM3A Regulatory Gene in Iranian Infertile Males. Iran. Biomed. J. 2019, 23, 220–227.
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301, doi:10.1056/NEJMoa0810069.
- Banka, S.; Lederer, D.; Benoit, V.; Jenkins, E.; Howard, E.; Bunstone, S.; Kerr, B.; McKee, S.; Lloyd, I.C.; Shears, D.; et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin. Genet. 2015, 87, 252–258, doi:10.1111/cge.12363.
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453, doi:10.1093/hmg/ddv180.
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124, doi:10.1016/j.ajhg.2011.11.021.
- Yang, P.; Tan, H.; Xia, Y.; Yu, Q.; Wei, X.; Guo, R.; Peng, Y.; Chen, C.; Li, H.; Mei, L.; et al. De novo exonic deletion of KDM6A in a Chinese girl with Kabuki syndrome: A case report and brief literature review. Am. J. Med. Genet. A 2016, 170, 1613–1621, doi:10.1002/ajmg.a.37634.
- Lindgren, A.M.; Hoyos, T.; Talkowski, M.E.; Hanscom, C.; Blumenthal, I.; Chiang, C.; Ernst, C.; Pereira, S.; Ordulu, Z.; Clericuzio, C.; et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum. Genet. 2013, 132, 537–552, doi:10.1007/s00439-013-1263-x.
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10, doi:10.1038/s41389-017-0011-9.
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17, doi:10.1038/s12276-019-0230-6.
- Kaelin, W.G., Jr. Cancer and altered metabolism: Potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 335–345, doi:10.1101/sqb.2011.76.010975.
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974, doi:10.1038/ncomms11974.
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5' UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010, doi:10.1016/j.cell.2015.10.012.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9, doi:10.1007/s00439-013-1358-4.
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621, doi:10.1038/ng1019.
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932, doi:10.1182/blood-2003-07-2535.
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290, doi:10.1371/journal.pmed.0030290.
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750, doi:10.1101/gad.276568.115.
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301, doi:10.1056/NEJMoa0810069.
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124, doi:10.1016/j.ajhg.2011.11.021.
- Yang, P.; Tan, H.; Xia, Y.; Yu, Q.; Wei, X.; Guo, R.; Peng, Y.; Chen, C.; Li, H.; Mei, L.; et al. De novo exonic deletion of KDM6A in a Chinese girl with Kabuki syndrome: A case report and brief literature review. Am. J. Med. Genet. A 2016, 170, 1613–1621, doi:10.1002/ajmg.a.37634.
- Lindgren, A.M.; Hoyos, T.; Talkowski, M.E.; Hanscom, C.; Blumenthal, I.; Chiang, C.; Ernst, C.; Pereira, S.; Ordulu, Z.; Clericuzio, C.; et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum. Genet. 2013, 132, 537–552, doi:10.1007/s00439-013-1263-x.
- Dunwoodie, S.L. The role of hypoxia in development of the Mammalian embryo. Dev Cell. 2009, 17, 755–773, doi:10.1016/j.devcel.2009.11.008.
- Banka, S.; Lederer, D.; Benoit, V.; Jenkins, E.; Howard, E.; Bunstone, S.; Kerr, B.; McKee, S.; Lloyd, I.C.; Shears, D.; et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin. Genet. 2015, 87, 252–258, doi:10.1111/cge.12363.
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453, doi:10.1093/hmg/ddv180.
- Li, S.; Ali, S.; Duan, X.; Liu, S.; Du, J.; Liu, C.; Dai, H.; Zhou, M.; Zhou, L.; Yang, L.; et al. JMJD1B Demethylates H4R3me2s and H3K9me2 to Facilitate Gene Expression for Development of Hematopoietic Stem and Progenitor Cells. Cell Rep. 2018, 23, 389–403, doi:10.1016/j.celrep.2018.03.051.
- Hojati, Z.; Soleimanpour, E.; Javadirad, S.M.; Nasr-Esfahani, M.H. Identification of Two Novel Mutations in KDM3A Regulatory Gene in Iranian Infertile Males. Iran. Biomed. J. 2019, 23, 220–227.
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10, doi:10.1038/s41389-017-0011-9.
- Kaelin, W.G., Jr. Cancer and altered metabolism: Potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 335–345, doi:10.1101/sqb.2011.76.010975.
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17, doi:10.1038/s12276-019-0230-6.
- Hancock, R.L.; Masson, N.; Dunne, K.; Flashman, E.; Kawamura, A. The Activity of JmjC Histone Lysine Demethylase KDM4A is Highly Sensitive to Oxygen Concentrations. ACS Chem. Biol. 2017, 12, 1011–1019, doi:10.1021/acschembio.6b00958.

