 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karol Bukowski | + 6159 word(s) | 6159 | 2020-05-06 09:50:25 | | | |
| 2 | Rita Xu | Meta information modification | 6159 | 2020-05-09 08:04:39 | | | | |
| 3 | Rita Xu | -21 word(s) | 6138 | 2020-10-28 10:58:59 | | |
Video Upload Options
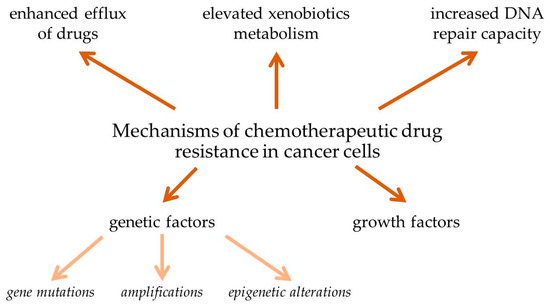
Cancer is one of the main causes of death worldwide. Despite the significant development of methods of cancer healing during the past decades, chemotherapy still remains the main method for cancer treatment. Multidrug resistance (MDR) is responsible for over 90% of deaths in cancer patients receiving traditional chemotherapeutics or novel targeted drugs. The mechanisms of MDR include elevated metabolism of xenobiotics, enhanced efflux of drugs, growth factors, increased DNA repair capacity, and genetic factors (gene mutations, amplifications, and epigenetic alterations). Rapidly increasing numbers of biomedical studies are focused on designing chemotherapeutics that are able to evade or reverse MDR.
1. Introduction
Statistical data shows that over 90% mortality of cancer patients is attributed to drug resistance. MDR of cancer cells during chemotherapy can be associated with a variety of mechanisms, including enhanced efflux of drugs, genetic factors (gene mutations, amplifications, and epigenetic alterations), growth factors, increased DNA repair capacity, and elevated metabolism of xenobiotics (Figure 1). Each of these mechanisms leads to reduction of the therapeutic efficacy of administered drugs, causing more difficulty in tumor treatment [1][2][3][4][5].
2. Enhanced Efflux of Drugs
ATP-binding cassette (ABC) proteins such as P-glycoprotein (P-gp)/ATP-binding cassette subfamily B member 1 (ABCB1) or Breast Cancer Resistance Protein (BCRP) present in the cell membrane are responsible for regulation of distribution, absorption, and excretion of a variety of chemical compounds. Because these proteins protect cells from death caused by high intracellular drug concentration, they can also interfere with drug administration, decreasing its bioavailability, intracellular concentration, and its transition of the blood–brain barrier (BBB). P-gp, highly expressed on the endothelial cell surface contributes to reduced chemotherapeutic drug penetration to the specific sites, especially in case of brain tumor treatment where anticancer agents are generally incapable of passing through the BBB. The size of the tumor also plays a crucial role in drug penetration. Because of the poor blood supply in large tumors, chemotherapeutic agents are usually less efficient in large tumors due to the poor blood supply compared to smaller ones with nearly unlimited access of oxygen and nutrient supply. The P-gp protects the brain from potentially damaging compounds but, at the same time, restricts access of therapeutic agents responsible for higher complexity of the therapy. In most cases, the only way to overcome the barrier is to increase the concentration of the drug, which often leads to systemic toxicity. This is the reason why elevated efflux of the drug has been considered to be one of the key mechanisms of cancer cell resistance against chemotherapeutics [1][3][4][6].
P-gp and BCRP can eliminate from cancer cells a wide variety of structurally and functionally unrelated anticancer agents, including epipodophyllotoxins, anthracyclines, vinca alkaloids, bisantrene, colchicine, taxanes, imatinib, saquinavir, camptothecins, thiopurines, actinomycin D, methotrexate, and mitoxantrone to the extracellular space, reducing intracellular drug accumulation [3][4][7][8][9]. Among a variety of chemotherapeutics, significant correlation between increased expression of P-gp in cancer cells and their enhanced resistance to paclitaxel, etoposide, olaparib, DOX, and vinblastine has been found [4][10][11][12][13]. Overexpression of P-gp has been observed in about 50% of all human cancers. While, in some tumor types such as lung, liver, kidney, rectum, and colon, increased P-gp expression has been observed before chemotherapy treatment, in others, including hematological malignancies such as acute lymphoblastic leukemia and acute myeloid leukemia, overexpression of P-gp has been noticed after anticancer agents exposure [4][9]. Overexpression of P-gp and BCRP has been associated with poor clinical response and MDR in patients with multiple myeloma, acute lymphocytic leukaemia, chronic lymphocytic leukaemia, acute myelogenous leukaemia, and metastatic breast cancer [7]. Additionally, it has been reported that P-gp plays a role in cancer cells MDR not only by participating in the efflux of intracellular chemotherapeutic agents but also by inhibiting tumor necrosis factor-related apoptosis-inducing ligand TRAIL-mediated and caspase-related pathways of apoptosis [8][14][15].
Although P-gp inhibitors show a high efficacy in vitro and in vivo studies, none of them have been approved by the U.S. Food and Drug Administration (FDA) for clinical use in cancer treatment [7][16]. However, Nanayakkara et al. [16] presented some new P-gp inhibitors, which potentially may be promising drugs in cancer chemotherapy. Despite the fact that they have still not entered clinical trials, researchers using computational approach found several compounds that were able to inhibit the P-gp activity and confirmed their anticancer properties against MDR cancer cell lines. Furthermore, Nanayakkara et al. [16] tested coadministration of chemotherapeutics with the investigated compounds against two-dimensional MDR prostate and ovarian cancer cells and three-dimensional prostate cancer microtumor spheroids. The results showed a significant decrease in cell motility and cell survival and viability. Additionally, the researchers demonstrated that all of the tested P-gp inhibitors did not exhibit toxic potential and were not P-gp transport substrates. Moreover, tested compounds increased not only cellular retention of anticancer agents but also the amount of reporter compounds being P-gp transport substrates.
Another example of new potential P-gp inhibitors are naturally occurring potassium ionophores such as salinomycin. Guberović et al. [17] demonstrated that some of the investigated crown ethers revealed to be significantly more efficient in sensitising MDR cells to adriamycin and paclitaxel compared to a well-known P-gp inhibitor verapamil.
Furthermore, the results obtained by Liu et al. [18] showed that combining administration of ascorbic acid with DOX could increase the sensitivity of human MDR breast cancer (MCF-7/MDR) cells to DOX in vitro and in vivo. As those researchers showed, ascorbate improved responsiveness of the cells to DOX through promoting the cellular accumulation of the drug associated with induction of reactive oxygen species-dependent ATP depletion.
Moreover, the compound that potentially could find its application in chemotherapy treatment is tometodione M (TTM), a novel natural syncarpic acid-conjugated monoterpene. In the study of Zhou et al. [19], the drug increased intracellular rhodamine 123 and DOX accumulation in human MDR leukemia cells (K562/MDR) and MCF-7/MDR cells by decreasing P-gp-related drug efflux. TTM reduced expression of both P-gp protein and mRNA via inhibition of p38 mitogen-activated protein kinase (MAPK) signaling, leading to MDR reversion in cancer cells. Additionally, TTM not only enhanced the cytotoxicity of docetaxel in K562/MDR and MCF-7/MDR cells but also triggered apoptosis and decreased colony formation in docetaxel-treated cells [19].
In addition, the results of Yuan et al. [20] demonstrated that cinobufagin, a substance isolated from the posterior auricular glands and skin of the Asiatic toad (Bufo gargarizans), affected modulation of P-gp activity in human P-gp-overexpressing colorectal carcinoma cells, including Caco-2/ADR, HCT116/L, and LoVo/ADR, which could potentially find it useful in combination with chemotherapeutic agents in colon cancer treatment. Data showed that cinobufagin significantly enhanced intracellular accumulation of rhodamine 123 and DOX and exhibited apoptotic properties in MDR cells. Moreover, cinobufagin remarkably influenced P-gp overexpressing in LoVo/ADR cells by increasing their sensitivity to DOX belonging to P-gp substrate drugs. Despite the fact that further investigations on the mechanisms of action of cinobufagin showed no changes in the expression of P-gp, a significant effect of cinobufagin on noncompetitive inhibition of P-gp ATPase activity was observed [20].
Furthermore, the chemical substance which could be potentially used in cancer chemotherapy is iso-pencillixanthone A (iso-PXA), which naturally occurs in the fungus Penicillium oxalicum. Chen et al. [21] found that iso-PXA could increase the intracellular concentration of (VCR) in the human cervical cancer cell line HeLa/VCR by P-gp ATPase stimulation and reduction in P-gp expression. As those researchers showed, iso-PXA effectively induced the intrinsic pathway of apoptosis by poly (ADP-ribose) polymerase (PARP), caspase-3, and caspase-9 activation. Moreover, iso-PXA initiated apoptotic events by degradation of induced myeloid leukemia cell differentiation protein (Mcl-1), accumulation of F-box and WD repeat domain-containing 7 protein (FBW7), and increase of the Bax/Bcl-2 ratio.
Chen et al. [22] investigated the association between the activities of natural flavonoids, including (−)-catechin, (−)-gallocatechin, luteolin, taxifolin, and human P-gp activity. The researchers found that taxifolin in a concentration-dependent manner significantly decreased ABCB1 expression and inhibited the P-gp function through DOX efflux and uncompetitive inhibition of rhodamine 123.
Quinidine is a well-known, FDA-approved drug used clinically for the treatment of pseudobulbar effect, arrhythmia, and malaria. However, side effects of the drug associated with myocardium condition, including factors such as torsade de pointes and long QT syndrome (LQTS), complicate its clinical usage as a P-gp inhibitor. The results of Snyder et al. [23] showed potential application of polymer-drug conjugates such as methoxypolyethylene glycol (mPEG) glycine-quinidine conjugate in reversing MDR through P-gp inhibition. The investigated conjugate not only inhibited the function of P-gp comparable to quinidine but also significantly mitigated distribution of quinidine into the mouse myocardium, resulting in reduced off-target pharmacologic effects.
Sitravatinib, a novel promising receptor tyrosine kinase inhibitor, which presently is on clinical trials, has been shown to be correlated with reversing MDR of P-gp and BCRP-overexpressing cancer cells. The investigated compound inhibited the drug efflux function of P-gp and BCRP in a concentration-dependent manner without altering the protein expression of P-gp and BCRP in MDR cancer cells. As Wu et al. [7] suggested, despite the fact that sitravatinib at submicromolar concentrations reversed MDR mediated by P-gp and BCRP, further clinical trials are required.
Furthermore, the effect of novel P-gp inhibitors, polyethylene glycol-modified titanium dioxide nanoparticles (TiO2 PEG NPs), on cisplatin cytotoxicity against P-gp overexpressing HepG2 cells was examined. This study showed that increased cisplatin cytotoxicity was associated with downregulation of the expression of P-gp in HepG2 cells by TiO2 PEG NPs [24].
3. Genetic Factors
3.1. Gene Mutations
Gene mutations, which are commonly observed in tumor cells are considered one of the main causes of the failure of chemotherapy treatment. As Duesberg et al. [25][26] concluded, the best explanation of MDR development in cancer cells is their aneuploidy nature. Researchers have suggested that frequently losing chromosomes or their reassortments during mitosis are responsible for losing drug-sensitive genes or for changes in biochemical pathways, which both seems to be crucial in chemotherapeutic drug resistance. In addition, normal cells, which rarely gain or lose a chromosome, usually stays sensitive to drugs, which makes the treatment even more complex.
Mutations of the TP53 gene, frequently observed in tumor cells, are one of the best-known biomarkers of the tumorigenesis. As Mantovani et al. [27] described, forty years of studies have demonstrated the irreplaceable role of the TP53 gene in protecting an organism against neoplastic transformation and tumor progression. The TP53 tumor suppressor is responsible for genome stability and cellular homeostasis by coordinating multiple processes and effector pathways, including regulation of the cell cycle and inducing apoptosis or G1 arrest in the case of any genotoxic stress caused during replication. Losing the tumor-suppressive activities by missense mutations in the TP53 gene, which are especially widespread in human cancers, reverses the protective role of the TP53 pathway by initiating chemoresistance, invasion, and metastasis. In a normal case, anticancer drugs, which induce DNA damage, cause cell death by TP53 activity. In contrast, loss of the TP53 activity in cancer cells allows continued replication no matter the type/level of DNA damage, which makes them resistant to genotoxic drugs (Figure 2) [27].
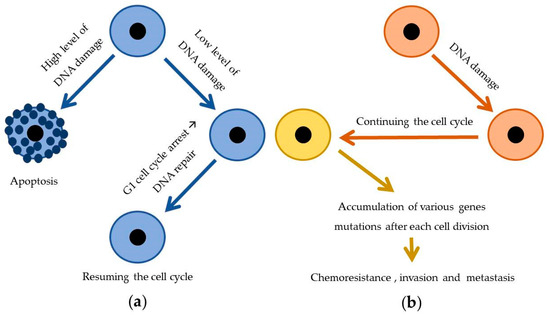
Figure 2. Differences in TP53 gene expression level between normal (a) and cancer cells (b) and consequences thereof: (a) accurate level of expression of TP53 gene and (b) decreased level of expression of TP53 gene [27].
Furthermore, function of the chimeric BCR-ABL gene seems to be key for initiation and maintenance of tumorigenicity, especially in chronic myeloid leukemia (CML). The oncogenic gene product increases frequency of cell division, blocks DNA repair, and inhibits apoptosis. BCR-ABL tyrosine-kinase inhibitors, such as imatinib, commonly used as the first-line drug for patients with CML, prevent ATP binding to the BCR-ABL kinase receptor, therefore inducing apoptosis in cancerous cells [28][29]. Data shows that mutations of the BCR-ABL gene associated with the drug-binding region commonly result in imatinib resistance during the CML treatment [28]. Additionally, in some clinical studies, scientists have observed significant correlation between reactivation of the BCR-ABL gene and remission of CML disease [30].
Topoisomerase II-targeted agents, such as etoposide, are frequently used in order to inhibit the replication process by stopping the activity of this enzyme. Unfortunately, gene mutations of topoisomerase alter its nuclear localization, which results in the tumor cells’ resistance to the use of drug. In addition, these drugs are not specific toward cancer cells, interacting with the entire genome, which significantly limits their safe usage in cancer management [31].
The aim of cytotoxic drugs is to disable components of cells, for which the functions are key for its survival. Because of the commonly observed gene mutations in tumor cells, they are able to make some alterations in response in target molecules, which make them resistant to the specific drug. The product of the mutated gene still retains its activity, but because of some changes in its stereochemical structure, it is not able to bind to the drug anymore. A well-known example of this mechanism of resistance is antiestrogen therapy of breast cancer. Patients, who initially show proper response to tamoxifen treatment, often at some point become insensitive to an endocrine manipulation. The state of complete unresponsiveness results from the gradual loss of estrogen receptors in mutated cells. Probably, an estrogen is no longer needed for growth and functioning of survived tumor cells [32][33].
3.2. Amplifications
The main role of many chemotherapeutics, such as methotrexate, is inhibition of key enzymes, e.g., dihydrofolate reductase engaged in controlling cell proliferation. Because of the possibility of gene amplification, which appears in 10% of the cancers, mainly in leukemias, cancer cells can overcome this inhibition by enhancing transcription of the gene, which encodes the enzyme. This process is associated with selective synthesis of a specific region of the chromosome, which provides multiple copies of the same gene. These amplified sequences are identified with homogeneously staining regions or double minute chromosomes. Each of those gene are transcribed in order to increase the mRNA level, which after that is used in the translation process to produce more enzymes. Because the drug concentration is limited, at some point, it is not able to inhibit the increased amount of enzyme [1][34].
Zhang et al. [35], using the clinically annotated genomic database, The Cancer Genome Atlas (TCGA), analyzed the transcriptomics, genomics, and clinical data of a variety of cancer samples, especially breast cancer (1082 samples). As the result, significant associations between amplification of the glycosylphosphatidylinositol-linked cell surface glycoprotein (CD24) gene and mutations in the TP53 gene, cancer proliferation, and metastasis were observed. As the researchers suggested, a copy number variation of CD24 could serve as a simple potential prognostic marker for identifying populations of interest for cancer treatment and risk subtype.
Other data present that factors such as gene rearrangements/amplification or anticancer drugs (e.g., rifampicin) could significantly increase the expression of ABCB1 gene, leading to elevation of P-gp activity. Data from one rifampicin therapy showed that the drug increased the intestinal P-gp level 3.5-fold and decreased the oral bioavailability of another used drug (digoxin) by 30% during the whole treatment [36][37].
Genovese et al. [38] observed that even application of a single dose of chemotherapeutics, such as DOX and paclitaxel targeting cancer cells lines or hematological malignancies and various solid tumors in vivo, led to amplification of chromosome region 7q21 containing gene ABCB1, subsequently resulting in overexpression of P-gp and other resistance-related proteins. As the researchers pointed out, additional factors such as epigenetic modifications and single nucleotide polymorphisms (SNPs) increased expression of ABCB1 as well. The results of a variety of studies have demonstrated that not only paclitaxel and DOX but also other anticancer agents, including anthracyclines and taxenes, caused overexpression and/or amplification of genes surrounding the ABCB1 locus.
The upregulation of oncogene human epidermal growth factor receptor-2 (HER2) expression occurs in approximately 20% of breast cancers. Amplification of HER2 leads to transcriptional modifications associated with a variety of genes and pathways in breast cancer cells. HER2 abnormal breast cancers are correlated with increased chemotherapy resistance and general worse prognosis. Anti-HER2 agents, such as lapatinib, trastuzumab, margetuximab, pertuzumab, and trastuzumab, have been used in HER2 abnormal breast cancers patients for many years. Unfortunately, administration of the inhibitor of HER2 signaling to HER2+ breast cancer patients often results in loss of initial drug sensitivity and development of resistance to used agent. The promising novel strategy of HER2 abnormal breast cancers’ clinical treatment assumes alternate combinatory agents, including cyclin-dependent kinase (CDK) 4/6 inhibitors, endocrine therapy, cholesterol pathway inhibitors, or receptor tyrosine kinase (RTK) inhibitors [39].
3.3. Epigenetic Alterations
The latest data strongly emphasizes the significant role of epigenetic alterations in cancer cells in anticancer drug resistance. Silencing tumor suppressor genes by their DNA hypermethylation or enhancing the expression of oncogenes by their DNA hypomethylation could be the potential factors involved in cancer development. During tumorigenesis, the epigenome goes through multiple alterations, including genome-wide loss of DNA methylation, regional hypermethylation (especially in CpG promoter islands of tumor suppressor genes), global changes in histone modification marks, and alterations in the miRNAs expression (Figure 3) [40][41][42].
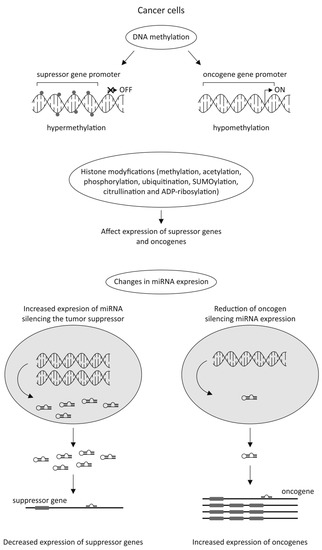
Currently, only two classes of epigenetic drugs have been approved by the FDA, i.e., DNA methylation inhibitors (iDNMT), including 5-azacitidine [43] and 5-aza-2′-deoxycytidine (decitabine; DAC) [44], as well as histone deacetylase inhibitors (iHDACs), such as Vorinostat, Belinostat, Romidepsin, and Panobinostat [45].
Demethylation of the ABCB1 gene in the cancer cell lines leads to decreased accumulation of the anticancer agent inside the cancer cells and results in acquisition of the MDR phenotype. Furthermore, the epigenetic alterations can affect the DNA repair system. For example, hypermethylation or mutation of the human MutL homolog 1 (hMLH1) gene, for which the product is involved in the mismatch repair system, can result in colorectal cancer development. Data has shown that the combination of conventional chemotherapeutics and epigenetic drugs such as DAC can be an effective solution in the treatment of cancerous cells and resisted tumors. Despite the fact that DAC does not affect directly the tumor growth, it inhibits DNA methylation which sensitizes the tumor to other chemotherapeutics, including carboplatin, cisplatin, and 5-FU [34].
Development of colorectal cancer is commonly associated with a variety of epigenetic alterations, such as histone modifications, DNA methylation, noncoding RNAs, and chromatin remodeling. While DNA methylation in genes MDF1, SSTR2, CMTM3, TGFB2, and NDRG4 is a potential marker for the detection of colorectal cancer in the early stages of its development, hypermetylation in gene CLDN11 is associated with metastasis and poor prospect of patient survival with colorectal cancer. Furthermore, silencing of tumor suppressor candidate 3 (TUSC3) mRNA expression by promoter methylation induces signaling of epidermal growth factor receptor (EGDR), which leads to colorectal cancer cells protection from apoptosis [46]. As Patnaik and Anupriya [46] suggested, development of DNA methyltransferase inhibitors and drugs targeting histone deacetylases could be a potential novel anticancer strategy. The latest data has demonstrated that CUDC-101 and CUDC-907, new synthesized histone deacetylase/kinase inhibitors, showed therapeutic potential as anticancer agents [47][48].
Despite the fact that microRNAs (miRNAs) have only 19–25 nucleotides and are unable to code any proteins, they affect regulation of gene expression by posttranscriptional modifications. Epigenetic changes associated with miRNAs frequently play an important role in the development of chemoresistance of various types of cancer. In recent years, many studies have shown that miRNAs affect the sensitivity of tumor cells against anticancer agents by influencing drug-resistance-related genes or genes related to cell proliferation, cell cycle, and apoptosis [49]. As Mansoori et al. [34] suggested, miRNAs could serve as a biomarker for prognosis of the effectiveness of chemotherapy treatment. The list of selected miRNAs involved in tumor transformation is presented in Table 1.
Table 1. Exemplary miRNAs that play an important role in cancer resistance.
| Cancer Type | miRNA | Chemotherapy Agent | Reference |
|---|---|---|---|
| prostate cancer | microRNA-34a | paclitaxel | [50] |
| microRNA-217, microRNA-181b-5p |
docetaxel, cabazitaxel | [51] | |
| pancreatic cancer | microRNA-320a micro-146 |
5-FU | [52] |
| microRNA-205, microRNA-7 | gemcitabine | [53] | |
| colorectal cancer | microRNA-519c | 5-FU | [54] |
| microRNAs-384 | oxaliplatin | [55] | |
| microRNA-96 | 5-FU | [56] | |
| cervical cancer | microRNA-499a | paclitaxel | [57] |
| microRNA -125a | paclitaxel | [58] | |
| microRNA-224 | paclitaxel | [59] | |
| breast cancer | microRNA-27b-3p | tamoxifen | [60] |
| microRNA-21 | trastuzumab | [61] | |
| microRNA-134 | DOX | [62] | |
| ovarian cancer | miR-23b | paclitaxel | [63] |
| microRNA-125b | paclitaxel | [64] | |
| microRNAs-449 | DOX | [65] | |
| gastric cancer | microRNA-508-5p | VCR, adriamycin, cisplatin, 5-FU |
[66] |
| microRNA-103/107 | DOX | [67] | |
| microRNA-495-3p | adriamycin, cisplatin, 5-FU, VCR | [68] |
4. Growth Factors
Experimental and clinical data have shown significant associations between inflammation and cancer development and progression. The results of accumulated experimental and clinical data have revealed that acute inflammation promotes tumor eradication while chronic immune response leads to tumor growth and invasion. Increased autocrine production of the growth factors, including interleukin (IL)-1, IL-4, IL-6, and IL-8, has been observed in MDR cancer cells, compared to drug-sensitive tumor cells [69][70][71][72].
It has been widely reported that IL-6 can affect various biological processes such as metabolism, differentiation, cell growth, and death by increasing ABCB1 gene expression and the CCAAT enhancer-binding protein family activation [70]. Furthermore, Ham et al. [72] provided convincing evidence for correlation between the activity of IL-6 in cancer-associated fibroblasts occurring in the tumor stroma and MDR of gastric cancer cells. The results of the researchers showed that IL-6 was a chromatin assembly factor-1 (CAF)-specific secretory protein, which conferred gastric cancer cell chemoresistance by paracrine signaling. Moreover, they observed that application of tocilizumab, an anti-IL-6 receptor monoclonal antibody, reversed the CAF-mediated inhibition of apoptosis in both in vitro and in vivo experimental models. This data demonstrated the potential therapeutic use of IL-6 inhibitors in order to increase the responsiveness to anticancer agents in gastric cancer cells.
The results of Wang et al. [70] indicated strong association between overexpression of IL-8 in tumor tissue, serum, ovarian cyst fluid, and ascites from ovarian cancer patients and poor sensitivity for a variety of anticancer agents used during their chemotherapy. As the researchers observed, MDR in ovarian cancer cells caused by increased expression of IL-8 was associated not only with activation of Ras/MEK/ERK and PI3K/Akt signaling and overexpression of MDR-related genes, including ABCB1 and apoptosis inhibitory proteins (XIAP, Bcl-xL, and Bcl-2) but also with decreased proteolytic activation of caspase-3. This is the reason why modulation of the IL-8 signaling pathway or IL-8 expression may be a potential strategy of MDR ovarian cancer treatment.
Cancer chemoresistance can be elevated not only by intracellular factors but also by increased level of extracellular fibroblast growth factors present in the media of metastatic and solid tumors. Data has shown that drugs with different mechanisms of action, including 5-FU, DOX, and paclitaxel, were ineffective against tumors with elevated levels of these extracellular factors. In order to prove the importance of fibroblast growth factors in development of cancer chemoresistance, Song et al. [73] applied suramin (a well-known inhibitor of these factors), which effectively reversed the 10-fold increased resistance observed in combination of intracellular and extracellular factors.
Glioblastoma, the most lethal brain cancer among adults, is a tumor characterized by marked genetic heterogeneity. However, changes in activation of receptor tyrosine kinase signaling are among the most common molecular modifications in glioblastoma. Data from a variety of studies has suggested significant association between signaling through fibroblast growth factor (FGF) receptors and glioblastoma progression. For that reason, blocking this signaling pathway by currently trialed small-molecule inhibitors of FGF receptors may be a potential strategy in glioblastoma treatment [74].
The aim of the study of Suzuki et al. [75] was to determine how extracellular FGFs affect the biology of small cell lung cancer (SCLC) cells and non-small cell lung cancer (NSCLC) cells. The results of the researchers showed significant associations between the activity of examined FGFs, i.e., FG2, FGF9, and FGF10, and proliferation, apoptosis, and treatment sensitivity of SCLC and NSCLC cells in vitro in a cell-specific manner.
Many data has shown that increased activity of protein kinase C [76] and extracellular matrix (ECM) [77] in breast tumor cells are associated with their chemotherapy resistance. It has been proven that ECM plays the key role in breast cancer progression, invasion, and metastasis. As Jena and Janjanam [77] suggested, remodeling of ECM is the major factor responsible for promoting cancer invasion and metastasis, especially matrix metalloproteinases (MMPs), including MMP-2, -9, -11, and -14, which degrade the matrix proteins. It was reported that β-D mannuronic acid could be a potential anticancer agent by inhibition of MMP-2 and -9. However, other factors such as ECM integrin b1-, b5-, and b6-; Hic-5 and ECM1 proteins; and enzymes, including heparanase, procollagen lysyl hydroxylase-2, LOXL2, and LOXL4 have also been shown to play a role in the regulation of breast cancer development and progression. Furthermore, stromal cells, including adipocytes, cancer-associated fibroblasts, and tumor-associated macrophages (TAMs) have been shown to be associated with tumor progression via a variety of processes (e.g., creating a vessel network which supports the nourishment of the tumor mass and secretion of vascular endothelial growth factor A (VEGF-A) by TAMs leading to tumor invasion) [77].
5. Increased DNA Repair Capacity
Another possibility of becoming tumor cells resistant to a variety of anticancer drugs is their ability to repair DNA damage. DNA repair endonuclease XPF and DNA excision repair protein ERCC1 involved in the nucleotide excision repair (NER) pathway are essential for the efficient repair of DNA damage induced by crosslinking and platinum-based agents [78]. A significant correlation between overexpression of both the XPF and ERCC-1 proteins and the development of cisplatin resistance in cancer cells was shown [79]. Low target specificity of a variety of anticancer drugs developed so far is the reason of their failure in chemotherapy treatment. However, successful use of PARP inhibitors against breast cancer (BRCA)-deficient tumors showed a new perspective on developing novel potential inhibitors of DNA repair proteins [80].
Novel compounds, i.e., E-X PPI2 and E-X AS7 have been identified as ERCC1-XPF inhibitors. Enhanced melanoma cell sensitivity to cisplatin, inhibition of NER activity, and decreased level of ERCC1-XPF heterodimers in ovarian cancer cells were observed after E-X PPI2 or E-X AS7 usage [81]. Additionally, one of the investigated catechol-based inhibitors of ERCC1-XPF (13 compound) displayed high activity in NER and selectivity against deoxyribonuclease I and Flap structure-specific endonuclease 1 (FEN-1), which resulted in enhanced cisplatin activity in A375 melanoma cells [82]. Gentile et al. [78], using a multistep computational approach, found potential modification sites of F06, an inhibitor of the ERCC1-XPF. Among investigated analogs of F06, increased IC50 value (0.49 µM) for the inhibition of ERCC1-XPF activity was observed in a case of B5 compound. These results require further testing and optimization; however, methods based on the computational approach described by the researchers can be used to develop novel potential ERCC1-XPF inhibitors.
Repair and tolerance of Pt-DNA lesions depend not only on NER but also on efficiency of homologous recombination (HR) pathway. Data has demonstrated that replication protein A (RPA) can be a new promising target in chemotherapy treatment. RPA, as a single-strand DNA (ssDNA)-binding protein, not only is involved in DNA recombination and replication but also plays key functions in DNA-damage response (DDR), HR, and NER DNA repair pathways [83][84][85]. In some studies, the activity of novel derivatives of RPA inhibitors against in vitro and in vivo models of NSCLC and epithelial ovarian cancer (EOC) has been examined. One of the investigated compounds, the TDRL-551, showed anticancer activity both as a single agent and in combination with Pt in an NSCLC in vivo model. In addition, synergy of TDRL-551 with platinum in both xenograft and tissue culture models of EOC was observed [84]. Furthermore, anticancer properties of one of the previously identified RPA inhibitors, SMI MCI13E, was investigated using both ovarian and lung cancer cell lines. In this study, decreasing RPA DNA-binding activity and disruption’s RPA role in the cell cycle regulation was noted. Addition of SMI MCI13E to cisplatin enhanced its anticancer properties. The results have shown that RPA small molecule inhibitors can be applied as a single chemotherapeutic or may be used in combination with current anticancer agents to enhance their efficacy [85]. Moreover, new analogues of a previously reported RPA inhibitor, TDRL-551, were designed in order to enhance physicochemical properties and anticancer activity. Compounds 43, 44, 45, and 46 were identified as chemical substances with increased solubility, low micromolar RPA inhibitory activity, and enhanced cellular uptake, holding promise for further investigation of novel chemotherapeutics.
Opposite to other DNA repair pathways, decreased activity of the DNA mismatch repair (MMR) pathway is associated with enhanced damage tolerance that leads to increased mutagenicity and chemoresistance. Hypermethylation of the hMLH1 gene promoter, causing a decreased expression of the MLH1 protein involved in the MMR pathway has been found in many cancers. In vitro studies have demonstrated that 5-fluoro-2-deoxycytidine and the cytidine analog, decitabine, can reverse this hypermethylation and increase cell sensitivity to cisplatin via restoring MMR functionality [86][87].
Ataxia telangiectasia and Rad3-related protein ATR kinase plays an essential role in the regulation of the DDR pathway. Its inhibition has been shown to be associated with enhanced sensitivity of some cancer cells in vitro to DNA-damaging agents, including platinum-based compounds. However, data about successful application of ATR inhibitors in vivo is limited. Hall et al. [88] examined VX-970 anticancer properties in both in vitro and in vivo lung cancer models. The researchers observed enhanced sensitivity of most of the investigated lung cancer cell lines in vitro to a variety of DNA-damaging drugs after VX-970 usage. In vivo, in primary lung xenografts derived from patients, VX-970 inhibited ATR activity in tumors and significantly increased the efficacy of cisplatin. The combination of cisplatin and VX-970 resulted in tumor regression in a model sensitive to cisplatin and complete inhibition of tumor growth in three cisplatin-resistance models [88]. The substance AZD6738 is another ATR kinase inhibitor which induces senescence and cell death in NSCLC cells. AZD6738 increases cytotoxicity of gemcitabine and cisplatin in NSCLC cell lines and enhances cisplatin anticancer properties in ATM-deficient NSCLC cells. ATR kinase inhibition caused by daily administration of AZD6738 for 14 days was well tolerated in mice and increased the therapeutic properties of cisplatin in xenograft models. The synergy of AZD6738 and cisplatin showed strong anticancer properties against ATM-deficient lung cancer xenografts [89].
Due to interrupted HR repair in BRCA1-deficient breast cancer cells, DNA double-strand breaks (DSBs) in these cells can be repaired only by the nonhomologous end joining (NHEJ) pathway. This is the reason why inhibition of DNA-dependent protein kinases (DNA-PKcs) involved in DDR and NHEJ pathways could be a new promising target in BRCA1-deficient breast cancer treatment [90][91]. Albarakati et al. [90] observed a synergy between cisplatin and two highly selective DNA-PKcs inhibitors, NU7026 and NU7441, in BRCA1-deficient breast cancer cell lines.
Sustained regressions in patient-derived xenograft models after treatment with AZD7648, the highly selective DNA-PK inhibitor and efficient sensitizer of DOX, and radiotherapy (radiation-induced DNA damage) was observed. In addition, combination of AZD7648 with olaparib, a well-known PARP inhibitor, resulted in cell growth inhibition, apoptosis, and enhanced genomic instability in ATM-deficient cells model. Furthermore, AZD7648 elevated olaparib efficacy in both patient-derived xenograft and xenograft models contributing to sustained tumor regression [91].
RAD51 is a protein involved in HR pathway responsible for DNA DSB repair. It binds to ssDNA and assists in HR repair by exchanging DNA strand breaks. Enhanced HR and overexpression of RAD51 have been found in multiple myeloma cells. Furthermore, high RAD51 expression in vivo has been shown to be correlated with chemoresistance and poor patient survival. The compound B02 interrupted binding RAD51 to ssDNA, inhibiting HR pathway, which resulted in enhanced cancer sensitivity to a variety of DNA damaging agents, such as DOX. In contrast, the combination of DOX and B02 had no impact on normal human CD19+ B cells from peripheral blood [92].
In a case of DNA-damaging chemotherapy treatment, the process of mutagenic translesion synthesis (TLS) was significantly associated with development of MDR in cancer cells. Wojtaszek et al. [93] demonstrated the highly specify small-molecule inhibitor JH-RE-06 that interrupted recruitment of mutagenic POL ζ involved in TLS activity. Coadministration of JH-RE with cisplatin increased cisplatin-induced cytotoxicity both in cultured mouse and human cell lines. Previous research also revealed association between disturbing POL ζ and enhanced efficiency of DNA-lesion chemotherapy [94].
The TLS pathway is responsible for repair of inter-strand DNA cross-links (ICLs). This process is regulated by Lys-164-mono-ubiquitinated proliferating cell nuclear antigen (PCNA). Inoue et al. [95] observed that T2 amino alcohol (T2AA) inhibited TLS repair and increased DNA DSBs by interrupting the function of Lys-164-mono-ubiquitinated PCNA. Blocking the interaction between genes involved in the DNA repair, REV1 and mono-ubiquitinated PCNA, resulted in inhibition of ICL repair and enhanced cisplatin cytotoxicity.
Mutation of TLS DNA polymerase Rev1 in cancer cells modified their TLS activity, increasing proliferating cells survival by enhancing tolerance to DNA damage occurring during replication. Sail et al. (2017) showed that two new synthesized compounds—4 and 5—inhibited mutagenic Rev1/Polζ-dependent TLS in cells, sensitizing human fibrosarcoma HT1080 cells to cisplatin. Additional experiments confirmed specificity of the investigated compounds, making them first inhibitors of TLS that target C-terminal domain of Rev1 (Rev1-CT) [96].
It has been reported that DNA DSBs in a DICER- and DROSHA-dependent manner generate DNA damage response RNAs (DDRNAs), responsible not only for the DDR management but also for guiding DNA repair. As Gioia et al. [97] observed, enoxacin, a compound being a DICER activity booster, increased the DDR signaling and DNA repair in cells exposed to ionizing radiations. Stimulation of DDRNAs production by enoxacin at dysfunctional telomeres and at chromosomal DSBs promoted accumulation of TP53 at damaged sites and, in consequence, caused suppression of homologous recombination, leading to DNA repair towards more accurate and faster nonhomologous end-joining. Unfortunately, augmented DNA repair elevated by the enoxacin not only increased the survival of normal cells but also affected cancer cells treated with anticancer agents, which might potentially result in acquisition of MDR phenotype by these cells.
6. Elevated Metabolism of Xenobiotics
As known, carrier molecules and enzymes responsible for drug metabolism play role in cancer cells chemotherapy resistance. Several studies have suggested that exposure to anticancer drugs may lead to induction and expression of gene products that protect the cell. Drug metabolizing enzymes are an integral part of phase I and phase II metabolism that helps in the detoxification of endogenous and exogenous substrates (xenobiotics).
Isoforms of cytochrome (CYP) such CYP1A6, CYP1A2, CYP1B1, CYP2C9, CYP2B6, CYP2C19, CYP3A4/5, and CYP2D6 are essential for phase I of drug metabolism and detoxification. Overexpression of CYP1B1 has been observed in various cancer cell types that modify the biotransformation of chemotherapeutics, such as mitoxantrone, flutamide, docetaxel, and paclitaxel [98]. Furthermore, increased expression of CYP2A6 enzyme, which is involved in metabolism of anticancer agents, including ifosfamide, cyclophosphamide, aflatoxin, and fluorouracil, has been reported in breast tumor tissues. In addition, highly upregulated expression of CP4Z1, CYP1B1, and CYP2A7 in cancer cells was associated with their enhanced resistance to a variety of chemotherapeutics [99].
Altered expression of enzymes involved in phase II of drug metabolism, including glutathione-S-transferases (GSTs), gamma-glutamyl transferases (γGTs), uridine diphospho-glucuronosyltransferases (UGTs), thiopurine methyltransferases (TPMTs), and dihydropyrimidine dehydrogenases (DPDs) in cancer cells may enhance their MDR [98][100]. The ability to inhibit the UGT activity, mainly UGT1A1, by kinase inhibitors, including sorafenib, regorafenib, pazopanib, and lapatinib, has been observed. However, in contrast to pazopanib and lapatinib activities, inhibition of UGT1A1 by sorafenib and regorafenib has been shown to be correlated with hyperbilirubinemia in patients [101]. Furthermore, novel UGT1A4 inhibitors that selectively increased sensitivity of cancer cells to chemotherapeutic agents demonstrated a new potential strategy in overcoming cancer MDR [102].
In order to overcome MDR in cancer cells with elevated GST and γGT expression, γGT-activated arsenic-based prodrugs, including darinaparsin and 4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid (GSAO), as well as GST-activated agents such as nitrogen mustard have been employed [100][103]. Additionally, natural flavonoids derivatives, such as phloretin, phloridzin, baicalein, and baicalin (with micromolar concentrations), were shown to be associated with inhibition of GST activity [104]. Other novel inhibitors of GST enzymes and chalcone derivatives, including 4-methoxychalcone, 4,4′-diflurochalcone, 2′-hydroxy-4-methoxychalcone, 4′-hydroxychalcone, and 4-fluorochalcone, were also reported [105].
As FeiFei et al. [106] found, there was a significant correlation between losing an F-box only protein 8 (FBX8), a key component of the SKP1-CUL1-F-box (SCF) E3 ubiquitin ligases, and acceleration of colon tumorigenesis. FBXB, through the ubiquitination process, led to degradation of GSTP1, resulting in suppression of colorectal cancer progression.
Glutathione (GSH) functions are associated with maintaining cellular redox homeostasis. GSH detoxifies xenobiotics as well as enhances MDR in cancer cells. In contrast to normal cells, cancer cells show a higher reactive oxygen species (ROS) production. Due to vicious proliferation and enhanced metabolism in cancer cells, these cells developed an enhanced antioxidant defense system to manage the elevated oxidant state. As many data suggested, alterations in GSH level have been correlated with multiple pathways of programmed cell death in cancer cells [107][108][109]. Tumor tissues derived from lung, liver, colon, and breast cancers show overexpression of GSH compared to normal tissues. The enhanced detoxifying ability of GSH in cancer cells has been shown to be associated with decreased activity of chemotherapeutic agents [107][109][110].
The impairment of the GSH antioxidant defense system could sensitize cancer cells to currently used chemotherapeutics. It was suggested that the moderate decline in the GSH level would be an effective strategy to increase the sensitivity of cancer cells to chemotherapies [111]. The ways to deplete the cellular GSH level include the following: reduction of GSH precursor availability [112][113], inhibition of the GSH synthesis process [114], increase of GSSG levels [115], direct conjugation with GSH [100], and promotion of cellular GSH efflux [116].
References
- Luqmani, Y.A.; Mechanisms of drug resistance in cancer chemotherapy. Medical Principles and Practice 2005, 14, 35–48, 10.1159/000086183.
- Qiong Wu; Zhiping Yang; Yongzhan Nie; Yongquan Shi; Daiming Fan; Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Letters 2014, 347, 159-166, 10.1016/j.canlet.2014.03.013.
- Jinglu Wang; Nicole Seebacher; Huirong Shi; QuanCheng Kan; Zhenfeng Duan; Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559-84571, 10.18632/oncotarget.19187.
- Xuan Wang; Haiyun Zhang; Xiaozhuo Chen; Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2019, 2, 141–160, 10.20517/cdr.2019.10.
- Sabrina Dallavalle; Vladimir Dobričić; Loretta Lazzarato; Elena Gazzano; Miguel Machuqueiro; Ilza Pajeva; Ivanka Tsakovska; Nace Zidar; Roberta Fruttero; Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resistance Updates 2020, 50, 100682, 10.1016/j.drup.2020.100682.
- Mesci, S.; Marakli, S.; Yazgan, B.; Yıldırım, T.; The effect of ATP-binding cassette (ABC) transporters in human cancers. International Journal of Science Letters 2019, 1, 1–14.
- Chung-Pu Wu; Sung-Han Hsiao; Yang-Hui Huang; Lang-Cheng Hung; Yi-Jou Yu; Yu-Tzu Chang; Tai-Ho Hung; Yu-Shan Wu; Sitravatinib Sensitizes ABCB1- and ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Drugs. Cancers 2020, 12, 195, 10.3390/cancers12010195.
- YaQiong Zu; Zhiyong Yang; Songshan Tang; Ying Han; Jun Ma; Effects of P-Glycoprotein and Its Inhibitors on Apoptosis in K562 Cells. Molecules 2014, 19, 13061-13075, 10.3390/molecules190913061.
- Serhan Karvar; The role of ABC transporters in anticancer drug transport. TURKISH JOURNAL OF BIOLOGY 2014, 38, 800-805, 10.3906/biy-1407-3.
- Jurjen S. Lagas; Lin Fan; Els Wagenaar; Maria L.H. Vlaming; Olaf Van Tellingen; Jos H. Beijnen; Alfred H. Schinkel; P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 Determine the Pharmacokinetics of Etoposide. Clinical Cancer Research 2009, 16, 130-140, 10.1158/1078-0432.ccr-09-1321.
- Suman Lal; Zee Wan Wong; Edwin Sandanaraj; Xiaoqiang Xiang; Peter Cher Siang Ang; Edmund J. D. Lee; Balram Chowbay; Influence of ABCB1 and ABCG2 polymorphisms on doxorubicin disposition in Asian breast cancer patients. Cancer Science 2008, 99, 816-823, 10.1111/j.1349-7006.2008.00744.x.
- Kazuhiro Satake; Megumi Tsukamoto; Yuji Mitani; Luis Octávio Regasini; Vanderlan S. Bolzani; Thomas Efferth; Hiroshi Nakagawa; Human ABCB1 confers cells resistance to cytotoxic guanidine alkaloids from Pterogyne nitens. Bio-Medical Materials and Engineering 2015, 25, 249-256, 10.3233/bme-151282.
- Aparajitha Vaidyanathan; Lynne Sawers; Anne-Louise Gannon; Probir Chakravarty; Alison L Scott; Susan E Bray; Michelle J Ferguson; Gillian Smith; ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. British Journal of Cancer 2016, 115, 431-441, 10.1038/bjc.2016.203.
- Paloma S. Souza; James P. Madigan; Jean-Pierre Gillet; Khyati Kapoor; Suresh V. Ambudkar; Raquel Ciuvalschi Maia; Michael M. Gottesman; King Leung Fung; Expression of the multidrug transporter P-glycoprotein is inversely related to that of apoptosis-associated endogenous TRAIL. Experimental Cell Research 2015, 336, 318-328, 10.1016/j.yexcr.2015.06.005.
- Hanan Galski; Tamar Oved-Gelber; Masha Simanovsky; Philip Lazarovici; Michael M. Gottesman; Arnon Nagler; P-glycoprotein-dependent resistance of cancer cells toward the extrinsic TRAIL apoptosis signaling pathway.. Biochemical Pharmacology 2013, 86, 584-96, 10.1016/j.bcp.2013.06.004.
- Amila K. Nanayakkara; Courtney A. Follit; Gang Chen; Noelle S. Williams; Pia D. Vogel; John G. Wise; Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Scientific Reports 2018, 8, 967, 10.1038/s41598-018-19325-x.
- Iva Guberović; Marko Marjanović; Marija Mioč; Katja Ester; Irena Martin-Kleiner; Tatjana Šumanovac Ramljak; Kata Mlinarić-Majerski; Marijeta Kralj; Crown ethers reverse P-glycoprotein-mediated multidrug resistance in cancer cells. Scientific Reports 2018, 8, 14467, 10.1038/s41598-018-32770-y.
- Yutong Liu; Ling Zhang; Zitai Ma; Li Tian; Yingchi Liu; Yuqing Liu; Qi Chen; Yanchun Li; Enlong Ma; Ascorbate promotes the cellular accumulation of doxorubicin and reverses the multidrug resistance in breast cancer cells by inducing ROS-dependent ATP depletion.. Free Radical Research 2019, 53, 758-767, 10.1080/10715762.2019.1628957.
- Xu-Wei Zhou; Yuan-Zheng Xia; Ya-Long Zhang; Jian-Guang Luo; Chao Han; Hao Zhang; Chao Zhang; Lei Yang; Ling-Yi Kong; Tomentodione M sensitizes multidrug resistant cancer cells by decreasing P-glycoprotein via inhibition of p38 MAPK signaling. Oncotarget 2017, 8, 101965-101983, 10.18632/oncotarget.21949.
- Zeting Yuan; Xiaojing Shi; Yanyan Qiu; Hui Yu; Xue He; Tingting Jia; Yu Zou; Cheng Liu; Ke Xu; Yanyan Qiu; et al. Reversal of P-gp-mediated multidrug resistance in colon cancer by cinobufagin. Oncology Reports 2017, 37, 1815-1825, 10.3892/or.2017.5410.
- Li Chen; Xinxin Li; Miaomiao Cheng; Siyuan Wang; Qiuhong Zheng; Qinying Liu; Iso-pencillixanthone A from a marine-derived fungus reverses multidrug resistance in cervical cancer cells through down-regulating P-gp and re-activating apoptosis. RSC Advances 2018, 8, 41192-41206, 10.1039/c8ra09506j.
- Hsiu-Ju Chen; Chia-Ying Li; Ying-Tzu Chang; Charles C.N. Wang; Hsiang-Yen Lee; Hui-Yi Lin; Chin-Chuan Hung; Taxifolin Resensitizes Multidrug Resistance Cancer Cells via Uncompetitive Inhibition of P-Glycoprotein Function.. Molecules 2018, 23, 3055, 10.3390/molecules23123055.
- Sarah Snyder; Shamanth Murundi; Lindsey Crawford; David Putnam; Enabling P-glycoprotein inhibition in multidrug resistant cancer through the reverse targeting of a quinidine-PEG conjugate.. Journal of Controlled Release 2019, 317, 291-299, 10.1016/j.jconrel.2019.11.027.
- Basma Salama; El-Said El-Sherbini; Gehad R. El-Sayed; Mohamed El-Adl; Koki Kanehira; Akiyoshi Taniguchi; The Effects of TiO2 Nanoparticles on Cisplatin Cytotoxicity in Cancer Cell Lines. International Journal of Molecular Sciences 2020, 21, 605, 10.3390/ijms21020605.
- Peter H. Duesberg; Reinhard Stindl; Rüediger Hehlmann; Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proceedings of the National Academy of Sciences 2000, 97, 14295-14300, 10.1073/pnas.97.26.14295.
- Peter Duesberg; Reinhard Stindl; Ruediger Hehlmann; Origin of multidrug resistance in cells with and without multidrug resistance genes: Chromosome reassortments catalyzed by aneuploidy. Proceedings of the National Academy of Sciences 2001, 98, 11283-11288, 10.1073/pnas.201398998.
- Fiamma Mantovani; Licio Collavin; Giannino Del Sal; Mutant p53 as a guardian of the cancer cell. Cell Death & Differentiation 2018, 26, 199-212, 10.1038/s41418-018-0246-9.
- Chodimella Chandrasekhar; Pasupuleti Santhosh Kumar; Potukuchi Venkata Gurunadha Krishna Sarma; Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Scientific Reports 2019, 9, 2412, 10.1038/s41598-019-38672-x.
- Graeme Greenfield; Ross McMullan; Nuala Robson; Julie McGimpsey; Mark A Catherwood; Mary Frances McMullin; Response to Imatinib therapy is inferior for e13a2 BCR-ABL1 transcript type in comparison to e14a2 transcript type in chronic myeloid leukaemia. BMC Hematology 2019, 19, 7, 10.1186/s12878-019-0139-2.
- Ya-Chen Tina Shih; Jorge E Cortes; Hagop M. Kantarjian; Treatment value of second-generation BCR-ABL1 tyrosine kinase inhibitors compared with imatinib to achieve treatment-free remission in patients with chronic myeloid leukaemia: a modelling study. The Lancet Haematology 2019, 6, e398-e408, 10.1016/s2352-3026(19)30087-0.
- Lorena Infante Lara; Sabine Fenner; Steven Ratcliffe; Albert Isidro-Llobet; Michael Hann; Ben Bax; Neil Osheroff; Coupling the core of the anticancer drug etoposide to an oligonucleotide induces topoisomerase II-mediated cleavage at specific DNA sequences.. Nucleic Acids Research 2018, 46, 2218-2233, 10.1093/nar/gky072.
- Susana M. Campos; Aromatase Inhibitors for Breast Cancer in Postmenopausal Women. The Oncologist 2004, 9, 126-136, 10.1634/theoncologist.9-2-126.
- John A. Katzenellenbogen; Christopher G. Mayne; Benita S. Katzenellenbogen; Geoffrey L Greene; Sarat Chandarlapaty; Structural Underpinnings of Estrogen Receptor Mutations in Endocrine Therapy Resistance. Nature Reviews Cancer 2018, 18, 377-388, 10.1038/s41568-018-0001-z.
- Behzad Mansoori; Ali Mohammadi; Sadaf Davudian; Solmaz Shirjang; Behzad Baradaran; The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Advanced Pharmaceutical Bulletin 2017, 7, 339-348, 10.15171/apb.2017.041.
- Peng Zhang; Pan Zheng; Yang Liu; Amplification of the CD24 Gene Is an Independent Predictor for Poor Prognosis of Breast Cancer.. Frontiers in Genetics 2019, 10, 560, 10.3389/fgene.2019.00560.
- Martin F. Fromm; Hans-Martin Kauffmann; Peter Fritz; Oliver Burk; Heyo K. Kroemer; Rolf W. Warzok; Michel Eichelbaum; Werner Siegmund; Dieter Schrenk; The Effect of Rifampin Treatment on Intestinal Expression of Human MRP Transporters. The American Journal of Pathology 2000, 157, 1575-1580, 10.1016/s0002-9440(10)64794-3.
- Bernd Greiner; Michel Eichelbaum; Péter Fritz; Hans-Peter Kreichgauer; Oliver Von Richter; Johannes Zundler; Heyo K. Kroemer; The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. Journal of Clinical Investigation 1999, 104, 147-153, 10.1172/jci6663.
- Ilaria Genovese; Andrea Ilari; Yehuda G. Assaraf; Francesco Fazi; Gianni Colotti; Not only P-glycoprotein: Amplification of the ABCB1- containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins. Drug Resistance Updates 2017, 32, 23-46, 10.1016/j.drup.2017.10.003.
- Reema Wahdan-Alaswad; Bolin Liu; Ann D. Thor; Targeted lapatinib anti-HER2/ErbB2 therapy resistance in breast cancer: opportunities to overcome a difficult problem. Cancer Drug Resistance 2020, 3, 1–20, 10.20517/cdr.2019.92.
- Zibo Zhao; Ali Shilatifard; Epigenetic modifications of histones in cancer. Genome Biology 2019, 20, 1-16, 10.1186/s13059-019-1870-5.
- Helai P. Mohammad; Olena Barbash; Caretha L. Creasy; Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nature Medicine 2019, 25, 403-418, 10.1038/s41591-019-0376-8.
- Helai P. Mohammad; Olena Barbash; Caretha L. Creasy; Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nature Medicine 2019, 25, 403-418, 10.1038/s41591-019-0376-8.
- E. Kaminskas; Ann T Farrell; Yong-Cheng Wang; Rajeshwari Sridhara; Richard Pazdur; FDA Drug Approval Summary: Azacitidine (5‐azacytidine, Vidaza ™ ) for Injectable Suspension. The Oncologist 2005, 10, 176-182, 10.1634/theoncologist.10-3-176.
- Pin-Fang He; Jing-Dong Zhou; Ng-Ming Yao; Ji-Chun Ma; Xiang-Mei Wen; Zhi-Hui Zhang; Xin-Yue Lian; Zi-Jun Xu; Jun Qian; Jiang Lin; et al. Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: A systematic review and meta-analysis. Oncotarget 2017, 8, 41498-41507, 10.18632/oncotarget.17241.
- Andrew Goey; Tristan M Sissung; Cody J Peer; William D. Figg; Pharmacogenomics and histone deacetylase inhibitors. Pharmacogenomics 2016, 17, 1807-1815, 10.2217/pgs-2016-0113.
- Srinivas Patnaik; Anupriya; Drugs Targeting Epigenetic Modifications and Plausible Therapeutic Strategies Against Colorectal Cancer.. Frontiers in Pharmacology 2019, 10, 588, 10.3389/fphar.2019.00588.
- Toshio Shimizu; Patricia M. Lorusso; Kyri P. Papadopoulos; Amita Patnaik; Muralidhar Beeram; Lon S. Smith; Drew W. Rasco; Theresa A. Mays; Glenda Chambers; Anna Ma; et al.Jing WangRobert LaliberteMaurizio VoiA. W. Tolcher Phase I First-in-Human Study of CUDC-101, a Multitargeted Inhibitor of HDACs, EGFR, and HER2 in Patients with Advanced Solid Tumors. Clinical Cancer Research 2014, 20, 5032-5040, 10.1158/1078-0432.ccr-14-0570.
- Seiichi Okabe; Yuko Tanaka; Mitsuru Moriyama; Akihiko Gotoh; Effect of dual inhibition of histone deacetylase and phosphatidylinositol-3 kinase in Philadelphia chromosome-positive leukemia cells. Cancer Chemotherapy and Pharmacology 2020, 85, 401-412, 10.1007/s00280-019-04022-x.
- Wengong Si; Jiaying Shen; Huilin Zheng; Weimin Fan; The role and mechanisms of action of microRNAs in cancer drug resistance. Clinical Epigenetics 2019, 11, 25, 10.1186/s13148-018-0587-8.
- Xiaobing Liu; Xing Luo; Yuqi Wu; Ding Xia; Wei Chen; Zhenqiang Fang; Jianping Deng; Yaxing Hao; Xia Yang; Teng Zhang; et al.Luqiang ZhouYingbing WuQingqing WangJie XuXiaoyan HuLongkun Li MicroRNA-34a Attenuates Paclitaxel Resistance in Prostate Cancer Cells via Direct Suppression of JAG1/Notch1 Axis. Cellular Physiology and Biochemistry 2018, 50, 261-276, 10.1159/000494004.
- Hui-Ming Lin; Iva Nikolic; Jessica Yang; Lesley Castillo; Niantao Deng; Chia-Ling Chan; Nicole K. Yeung; Eoin Dodson; Benjamin Elsworth; Calan Spielman; et al.Brian Y. LeeZoë BoyerKaylene SimpsonRoger J. DalyLisa G. HorvathAlexander Swarbrick MicroRNAs as potential therapeutics to enhance chemosensitivity in advanced prostate cancer.. Scientific Reports 2018, 8, 7820, 10.1038/s41598-018-26050-y.
- Weibin Wang; Lijun Zhao; Xueju Wei; Lanlan Wang; Siqi Liu; Yu Yang; Fang Wang; Guotao Sun; Junwu Zhang; Yanni Ma; et al.Yupei ZhaoJia Yu MicroRNA-320a promotes 5-FU resistance in human pancreatic cancer cells. Scientific Reports 2016, 6, 27641, 10.1038/srep27641.
- Saurabh Singh; Deepak Chitkara; Virender Kumar; Stephen W. Behrman; Ram I. Mahato; miRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Letters 2013, 334, 211-220, 10.1016/j.canlet.2012.10.008.
- Kenneth K.W. To; W.W. Leung; Simon Siu-Man Ng; Exploiting a novel miR-519c–HuR–ABCG2 regulatory pathway to overcome chemoresistance in colorectal cancer. Experimental Cell Research 2015, 338, 222-231, 10.1016/j.yexcr.2015.09.011.
- Jasmine Evert; Surajit Pathak; Xiao-Feng Sun; Hong Zhang; A Study on Effect of Oxaliplatin in MicroRNA Expression in Human Colon Cancer. Journal of Cancer 2018, 9, 2046-2053, 10.7150/jca.24474.
- Sun-Ah Kim; Injung Kim; Sungjoo Kim Yoon; Eun Kyung Lee; Hyo‐Jeong Kuh; Indirect modulation of sensitivity to 5-fluorouracil by microRNA-96 in human colorectal cancer cells. Archives of Pharmacal Research 2014, 38, 239-248, 10.1007/s12272-014-0528-9.
- Yibing Chen; Yucen Song; Yanjun Mi; Huan Jin; Jun Cao; Haolong Li; Liping Han; Ting Huang; Xiaofei Zhang; Shumin Ren; et al.Qian MaZhengzhi Zou microRNA-499a promotes the progression and chemoresistance of cervical cancer cells by targeting SOX6. Apoptosis 2020, 25, 205-216, 10.1007/s10495-019-01588-y.
- Z Fan; H Cui; H Yu; Q Ji; L Kang; B Han; J Wang; Q Dong; Y Li; Z Yan; et al.X YanX ZhangZ LinY HuS Jiao MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression.. Oncogenesis 2016, 5, e197-e197, 10.1038/oncsis.2016.1.
- F Lin; P Wang; Y Shen; X Xie; Upregulation of microRNA-224 sensitizes human cervical cells SiHa to paclitaxel.. European Journal of Gynaecological Oncology 2015, 36, 432–436.
- Jiang Zhu; Zhengzhi Zou; Peipei Nie; Xiaoni Kou; Baoyan Wu; Songmao Wang; Zhangjun Song; Jianjun He; Downregulation of microRNA-27b-3p enhances tamoxifen resistance in breast cancer by increasing NR5A2 and CREB1 expression.. Cell Death & Disease 2016, 7, e2454-e2454, 10.1038/cddis.2016.361.
- Leticia De Mattos-Arruda; Giulia Bottai; Paolo Nuciforo; Luca Di Tommaso; Elisa Giovannetti; Vicente Peg; Agnese Losurdo; José Perez-Garcia; Giovanna Masci; Fabio Corsi; et al.Javier CortesJoan SeoaneGeorge Adrian CalinLibero Santarpia MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget 2015, 6, 37269-37280, 10.18632/oncotarget.5495.
- Lin Lu; Fang Ju; Hui Zhao; Xuezhen Ma; MicroRNA-134 modulates resistance to doxorubicin in human breast cancer cells by downregulating ABCC1. Biotechnology Letters 2015, 37, 2387-2394, 10.1007/s10529-015-1941-y.
- Jing Yan; Jing-Yi Jiang; Xiao-Na Meng; Yin-Ling Xiu; Zhi-Hong Zong; MiR-23b targets cyclin G1 and suppresses ovarian cancer tumorigenesis and progression.. Journal of Experimental & Clinical Cancer Research 2016, 35, 31, 10.1186/s13046-016-0307-1.
- Xiaoyan Ying; Kuang Wei; Zhe Lin; Yugui Cui; Jie Ding; Yun Chen; Boqun Xu; MicroRNA-125b Suppresses Ovarian Cancer Progression via Suppression of the Epithelial-Mesenchymal Transition Pathway by Targeting the SET Protein. Cellular Physiology and Biochemistry 2016, 39, 501-510, 10.1159/000445642.
- Eduardo Tormo; Sandra Ballester; Anna Adam-Artigues; Octavio Burgués; Elisa Alonso; Begoña Bermejo; Silvia Menéndez; Sandra Zazo; Juan Madoz-Gúrpide; Ana Rovira; et al.Joan AlbanellFederico RojoAna LluchPilar Eroles The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Scientific Reports 2019, 9, 5316, 10.1038/s41598-019-41472-y.
- Y Shang; Z Zhang; Z Liu; B Feng; G Ren; K Li; L Zhou; Y Sun; M Li; J Zhou; et al.Y AnK WuY NieD Fan miR-508-5p regulates multidrug resistance of gastric cancer by targeting ABCB1 and ZNRD1. Oncogene 2013, 33, 3267-3276, 10.1038/onc.2013.297.
- Zhiyu Wang; Neng Wang; Pengxi Liu; Qianjun Chen; Honglin Situ; Ting Xie; Jianxing Zhang; Cheng Peng; Yi Lin; Jianping Chen; et al. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget 2014, 5, 7013-7026, 10.18632/oncotarget.2192.
- Sheng Chen; Jian Wu; Kai Jiao; Qiong Wu; Jiaojiao Ma; Di Chen; Jianqin Kang; Guodong Zhao; Yongquan Shi; Daiming Fan; et al.Guohong Zhao MicroRNA-495-3p inhibits multidrug resistance by modulating autophagy through GRP78/mTOR axis in gastric cancer. Cell Death & Disease 2018, 9, 1070, 10.1038/s41419-018-0950-x.
- Sarra Setrerrahmane; Hanmei Xu; Tumor-related interleukins: old validated targets for new anti-cancer drug development. Molecular Cancer 2017, 16, 153, 10.1186/s12943-017-0721-9.
- Yue Wang; Ye Qu; Xiu Long Niu; Wei Jia Sun; Xiao Lei Zhang; Ling Zhi Li; Autocrine production of interleukin-8 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cytokine 2011, 56, 365-375, 10.1016/j.cyto.2011.06.005.
- D Conze; L Weiss; P S Regen; A Bhushan; D Weaver; P Johnson; Mercedes Rincon; Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells.. Cancer Research 2001, 61, 8851–8858.
- In-Hye Ham; Hye Jeong Oh; Hyejin Jin; Cheong A Bae; Sang-Min Jeon; Kyeong Choi; Sang-Yong Son; Sang-Uk Han; Rolf A. Brekken; Dakeun Lee; et al.Hoon Hur Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Molecular Cancer 2019, 18, 68, 10.1186/s12943-019-0972-8.
- SaeHeum Song; M. Guillaume Wientjes; Yuebo Gan; Jessie L.-S. Au; Fibroblast growth factors: An epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proceedings of the National Academy of Sciences 2000, 97, 8658-8663, 10.1073/pnas.140210697.
- Ana Jimenez-Pascual; Florian A Siebzehnrubl; Fibroblast Growth Factor Receptor Functions in Glioblastoma.. Cells 2019, 8, 715, 10.3390/cells8070715.
- Takahiro Suzuki; Hiroyuki Yasuda; Koji Funaishi; Daisuke Arai; Kota Ishioka; Keiko Ohgino; Tetsuo Tani; Junko Hamamoto; Ayano Ohashi; Katsuhiko Naoki; et al.Tomoko BetsuyakuKenzo Soejima Multiple roles of extracellular fibroblast growth factors in lung cancer cells. International Journal of Oncology 2014, 46, 423-429, 10.3892/ijo.2014.2718.
- Rishi Kant Singh; Sanjay Kumar; Pramod Kumar Gautam; Munendra Singh Tomar; Praveen Kumar Verma; Surya Pratap Singh; Arbind Acharya; Protein kinase C-α and the regulation of diverse cell responses. Biomolecular Concepts 2017, 8, 143-153, 10.1515/bmc-2017-0005.
- Manoj K. Jena; Jagadeesh Janjanam; Role of extracellular matrix in breast cancer development: a brief update. F1000Research 2018, 7, 274, 10.12688/f1000research.14133.2.
- Francesco Gentile; Ahmed H. Elmenoufy; Gloria Ciniero; David Jay; Feridoun Karimi‐Busheri; Khaled H. Barakat; Michael Weinfeld; Frederick G. West; Jack A. Tuszyński; Computer‐aided drug design of small molecule inhibitors of the ERCC1‐XPF protein–protein interaction. Chemical Biology & Drug Design 2020, 95, 460-471, 10.1111/cbdd.13660.
- Rafael Rosell; Miquel Tarón; Aurelio Ariza; Agustí Barnadas; Jose Luis Mate; Noemı́ Reguart; Mireia Margelı́; Enriqueta Felip; Pedro Mendez; Rosario Garcı́a-Campelo; et al. Molecular predictors of response to chemotherapy in lung cancer. Seminars in Oncology 2004, 31, 20-27, 10.1053/j.seminoncol.2003.12.011.
- Clarissa Ribeiro Reily Rocha; Matheus Molina Silva; Annabel Quinet; Januario Bispo Cabral-Neto; Carlos Fm Menck; DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics 2018, 73, e478s, 10.6061/clinics/2018/e478s.
- Ewan M. McNeil; Katy R. Astell; Ann-Marie Ritchie; Steven Shave; Douglas R. Houston; Preeti Bakrania; Hayley M. Jones; Puneet Khurana; Claire Wallace; Tim Chapman; et al.Martin A. WearMalcolm D. WalkinshawBarbara SaxtyDavid W. Melton Inhibition of the ERCC1–XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19-28, 10.1016/j.dnarep.2015.04.002.
- Timothy M. Chapman; Kevin J. Gillen; Claire Wallace; Maximillian Lee; Preeti Bakrania; Puneet Khurana; Peter J. Coombs; Laura Stennett; Simon Fox; Emilie A. Bureau; et al.Janet BrownleesDavid W. MeltonBarbara Saxty Catechols and 3-hydroxypyridones as inhibitors of the DNA repair complex ERCC1-XPF. Bioorganic & Medicinal Chemistry Letters 2015, 25, 4097-4103, 10.1016/j.bmcl.2015.08.031.
- Akaash K. Mishra; Silvana S. Dormi; Alaina M. Turchi; Derek S. Woods; John J. Turchi; Chemical inhibitor targeting the replication protein A-DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer.. Biochemical Pharmacology 2014, 93, 25-33, 10.1016/j.bcp.2014.10.013.
- Tracy M. Neher; Diane Bodenmiller; Richard W. Fitch; Shadia I. Jalal; John J. Turchi; Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin.. Molecular Cancer Therapeutics 2011, 10, 1796-806, 10.1158/1535-7163.MCT-11-0303.
- Navnath, S.; Gavande, N.S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Vernon, T.L.; Jordan, M.R.; Turchi, J.J. Structure-Guided Optimization of Replication Protein A (RPA)–DNA Interaction Inhibitors. ACS Med. Chem.Lett. 2020.
- Mark R. Kelley; Derek Logsdon; Melissa Fishel; Targeting DNA repair pathways for cancer treatment: what’s new?. Future Oncology 2014, 10, 1215-1237, 10.2217/fon.14.60.
- Thomas Helleday; Eva Petermann; Cecilia Lundin; Ben Hodgson; Ricky A. Sharma; DNA repair pathways as targets for cancer therapy. Nature Reviews Cancer 2008, 8, 193-204, 10.1038/nrc2342.
- Amy B. Hall; Dave Newsome; Yuxin Wang; Diane M. Boucher; Brenda Eustace; Yong Gu; Brian Hare; Mac A. Johnson; Sean Milton; Cheryl E. Murphy; et al.Darin TakemotoCrystal TolmanMark WoodPeter CharltonJean-Damien CharrierBrinley FureyJulian GolecPhilip M. ReaperJohn R. Pollard Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674-5685, 10.18632/oncotarget.2158.
- Frank P. Vendetti; Alan Lau; Sandra Schamus; Thomas P. Conrads; Mark J. O’Connor; Christopher J. Bakkenist; The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289-44305, 10.18632/oncotarget.6247.
- Nada AlBarakati; Tarek M.A. Abdel-Fatah; Rachel Doherty; Roslin Russell; Devika Agarwal; Paul Moseley; Christina Perry; Arvind Arora; Nouf AlSubhi; Claire Seedhouse; et al.Emad A. RakhaAndrew R. GreenGraham BallStephen ChanCarlos CaldasIan EllisSrinivasan Madhusudan Targeting BRCA1‐BER deficient breast cancer by ATM or DNA‐PKcs blockade either alone or in combination with cisplatin for personalized therapy. Molecular Oncology 2014, 9, 204-217, 10.1016/j.molonc.2014.08.001.
- Jacqueline H. L. Fok; Antonio Ramos-Montoya; Mercedes Vazquez-Chantada; Paul W. G. Wijnhoven; Valeria Follia; Neil James; Paul M. Farrington; Ankur Karmokar; Sophie E. Willis; Jonathan Cairns; et al.Jenni NikkiläDavid BeattieGillian M. LamontM. Raymond V. FinlayJoanne WilsonAaron SmithLenka Oplustil O’ConnorStephanie LingStephen E. FawellMark J. O’ConnorSimon J. HollingsworthEmma DeanFrederick W. GoldbergBarry R. DaviesElaine B. Cadogan AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity.. Nature Communications 2019, 10, 5065-15, 10.1038/s41467-019-12836-9.
- David A. Alagpulinsa; Srinivas Ayyadevara; Robert Joseph Shmookler Reis; A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Frontiers in Oncology 2014, 4, 289, 10.3389/fonc.2014.00289.
- Jessica L. Wojtaszek; Nimrat Chatterjee; Javaria Najeeb; Azucena Ramos; Minhee Lee; Ke Bian; Jenny Xue; Benjamin A. Fenton; Hyeri Park; Deyu Li; et al.Michael T. HemannJiyong HongGraham C. WalkerPei Zhou A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152-159.e11, 10.1016/j.cell.2019.05.028.
- Kinrin Yamanaka; Nimrat Chatterjee; Michael T. Hemann; Graham C. Walker; Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy?. PLOS Genetics 2017, 13, e1006842, 10.1371/journal.pgen.1006842.
- Akira Inoue; Sotaro Kikuchi; Asami Hishiki; Youming Shao; Richard Heath; Benjamin J. Evison; Marcelo Actis; Christine E. Canman; Hiroshi Hashimoto; Naoaki Fujii; et al. A Small Molecule Inhibitor of Monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) Inhibits Repair of Interstrand DNA Cross-link, Enhances DNA Double Strand Break, and Sensitizes Cancer Cells to Cisplatin. Journal of Biological Chemistry 2014, 289, 7109-7120, 10.1074/jbc.m113.520429.
- Vibhavari Sail; Alessandro A. Rizzo; Nimrat Chatterjee; Radha Charan Dash; Zuleyha Ozen; Graham C. Walker; Dmitry M. Korzhnev; M. Kyle Hadden; Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein−Protein Interaction. ACS Chemical Biology 2017, 12, 1903-1912, 10.1021/acschembio.6b01144.
- Ubaldo Gioia; Sofia Francia; Matteo Cabrini; Silvia Brambillasca; Flavia Michelini; Corey W. Jones-Weinert; Fabrizio Daddadifagagna; Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs.. Scientific Reports 2019, 9, 6460, 10.1038/s41598-019-42892-6.
- Shelly Pathania; Rohit Bhatia; Ashish Baldi; Randhir Singh; Ravindra K. Rawal; Drug metabolizing enzymes and their inhibitors' role in cancer resistance. Biomedicine & Pharmacotherapy 2018, 105, 53-65, 10.1016/j.biopha.2018.05.117.
- Yan Li; Albert Steppi; Yidong Zhou; Feng Mao; Philip Craig Miller; Max M. He; Tingting Zhao; Qiang Sun; Jinfeng Zhang; Tumoral expression of drug and xenobiotic metabolizing enzymes in breast cancer patients of different ethnicities with implications to personalized medicine.. Scientific Reports 2017, 7, 4747, 10.1038/s41598-017-04250-2.
- Emma Ramsay; Pierre J Dilda; Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Frontiers in Pharmacology 2014, 5, 181, 10.3389/fphar.2014.00181.
- John O. Miners; Nuy Chau; Andrew Rowland; Kushari Burns; Ross McKinnon; Peter I. MacKenzie; Geoffrey T Tucker; Kathleen M Knights; Ganessan Kichenadasse; Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: Implications for hyperbilirubinemia. Biochemical Pharmacology 2017, 129, 85-95, 10.1016/j.bcp.2017.01.002.
- Michael J. Osborne; Luciana Coutinho De Oliveira; Laurent Volpon; Hiba Ahmad Zahreddine; Katherine L. B. Borden; Overcoming Drug Resistance through the Development of Selective Inhibitors of UDP-Glucuronosyltransferase Enzymes. Journal of Molecular Biology 2019, 431, 258-272, 10.1016/j.jmb.2018.11.007.
- Jennifer Wu; Charles Henderson; Lynn Feun; Peter Van Veldhuizen; Philip Gold; Hui Zheng; Theresa Ryan; Lawrence S. Blaszkowsky; Haobin Chen; Max Costa; et al.Barry RosenzweigMarylynn NierodzikHoward HochsterFranco MuggiaGiovanni AbbadessaJonathan LewisAndrew X. Zhu Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Investigational New Drugs 2009, 28, 670-676, 10.1007/s10637-009-9286-9.
- Mine Aksoy; Irfan Küfrevioglu; Inhibition of human erythrocyte glutathione S-transferase by some flavonoid derivatives. Toxin Reviews 2017, 37, 251-257, 10.1080/15569543.2017.1345945.
- Muhammet Serhat Özaslan; Yeliz Demir; Hatice Esra Aslan; Şükrü Beydemir; Ömer Irfan Küfrevioğlu; Evaluation of chalcones as inhibitors of glutathione S-transferase. Journal of Biochemical and Molecular Toxicology 2018, 32, e22047, 10.1002/jbt.22047.
- Wang Feifei; Xu Honghai; Yan Yongrong; Wu Pingxiang; Wu Jianhua; Zhu Xiaohui; Li Jiaoying; Sun Jingbo; Zhou Kun; Ren Xiaoli; et al.Qi LuLan XiaoliangCheng ZhiqiangTang NaLiao WentingYanqing DingLiang Li FBX8 degrades GSTP1 through ubiquitination to suppress colorectal cancer progression. Cell Death & Disease 2019, 10, 351, 10.1038/s41419-019-1588-z.
- Huanhuan Lv; Chenxiao Zhen; Junyu Liu; Pengfei Yang; Lijiang Hu; Peng Shang; Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy.. Oxidative Medicine and Cellular Longevity 2019, 2019, 3150145-16, 10.1155/2019/3150145.
- Sofia C. Nunes; Jacinta Serpa; Glutathione in Ovarian Cancer: A Double-Edged Sword. International Journal of Molecular Sciences 2018, 19, 1882, 10.3390/ijms19071882.
- Ankita Bansal; M. Celeste Simon; Glutathione metabolism in cancer progression and treatment resistance. Journal of Cell Biology 2018, 217, 2291-2298, 10.1083/jcb.201804161.
- Enrico Desideri; Fabio Ciccarone; Maria Rosa Ciriolo; Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy.. Nutrients 2019, 11, 1926, 10.3390/nu11081926.
- Hideaki Ogiwara; Kazuaki Takahashi; Mariko Sasaki; Takafumi Kuroda; Hiroshi Yoshida; Reiko Watanabe; Ami Maruyama; Hideki Makinoshima; Fumiko Chiwaki; Hiroki Sasaki; et al.Tomoyasu KatoAikou OkamotoTakashi Kohno Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177-190.e8, 10.1016/j.ccell.2018.12.009.
- Young-Sun Lee; Dae-Hee Lee; So Yeon Jeong; Seong Hye Park; Sang Cheul Oh; Yong Seok Park; Jian Yu; Haroon A Choudry; David L Bartlett; Yong J. Lee; et al. Ferroptosis‐inducing agents enhance TRAIL‐induced apoptosis through upregulation of death receptor 5. Journal of Cellular Biochemistry 2018, 120, 928-939, 10.1002/jcb.27456.
- Xiaofen Pan; Zhixiu Lin; Danxian Jiang; Ying Yu; Donghong Yang; Hechao Zhou; Dechao Zhan; Sha Liu; Gang Peng; Zihong Chen; et al.Zhong-Hua Yu Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncology Letters 2019, 17, 3001-3008, 10.3892/ol.2019.9888.
- Judith Villablanca; Samuel L. Volchenboum; Hwangeui Cho; Min H. Kang; Susan L. Cohn; Clarke P. Anderson; Araz Marachelian; Susan Groshen; Denice Tsao-Wei; Katherine Matthay; et al.John M. MarisCharlotte E. HasenauerScarlett CzarneckiHollie LaiFariba GoodarzianHiro ShimadaC. Patrick Reynolds A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatric Blood & Cancer 2016, 63, 1349-1356, 10.1002/pbc.25994.
- Daniela Catanzaro; Edoardo Gaude; Genny Orso; Carla Giordano; Giulia Guzzo; Andrea Rasola; Eugenio Ragazzi; Laura Caparrotta; Christian Frezza; Monica Montopoli; et al. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102-30114, 10.18632/oncotarget.4945.
- Mohamed Elgendy; Marco Cirò; Amir Hosseini; Jakob Weiszmann; Luca Mazzarella; Elisa Ferrari; Riccardo Cazzoli; G. Curigliano; Andrea DeCensi; Bernardo Bonanni; et al.Alfredo BudillonPier Giuseppe PelicciVeerle JanssensManfred OgrisManuela BaccariniLuisa LanfranconeWolfram WeckwerthMarco FoianiSaverio Minucci Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798-815.e5, 10.1016/j.ccell.2019.03.007.


