 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paolo Iadarola | + 1431 word(s) | 1431 | 2021-02-19 09:10:04 | | | |
| 2 | Lily Guo | Meta information modification | 1431 | 2021-02-25 07:30:44 | | |
Video Upload Options
Alpha-1 antitrypsin (AAT) is a protein produced by the liver and released into the blood. As a known genetic cause of chronic obstructive pulmonary disease (COPD), alpha1-antitrypsin deficiency (AATD) can cause severe respiratory problems at a relatively young age. These problems are caused by decreased or absent levels of alpha1-antitrypsin (AAT), an antiprotease which is primarily functional in the respiratory system. If the levels of AAT fall below the protective threshold of 11 µM, the neutrophil-derived serine proteases neutrophil elastase (NE) and proteinase 3 (PR3), which are targets of AAT, are not sufficiently inhibited, resulting in excessive degradation of the lung parenchyma, increased inflammation, and increased susceptibility to infections. Because other therapies are still in the early phases of development, the only therapy currently available for AATD is AAT augmentation therapy. The controversy surrounding AAT augmentation therapy concerns its efficiency, as protection of lung function decline is not demonstrated, despite the treatment’s proven significant effect on lung density change in the long term.
1. Introduction
Despite thousands of people in the world being diagnosed with the autosomal co-dominant disease alpha1-antitrypsin deficiency (AATD), this condition is estimated to be highly underdiagnosed; the low awareness of physicians is perceived as the greatest barrier to the diagnosis rate[1]. AATD is characterised by intra- and extra-pulmonary diseases. The intra-pulmonary diseases include lung emphysema, bronchiectasis, and chronic bronchitis. Due to the resulting fixed airflow obstruction, most individuals with AATD experience symptoms of dyspnoea, exercise intolerance, and fatigue. Furthermore, these patients are more prone to infections, and inflammation is normally present in their lung tissue. The extrapulmonary diseases include liver dysfunction, panniculitis, granulomatosis with polyangiitis (GPA), and ulcerative colitis, the latter of which occur only rarely. Based on the irreversible airflow obstruction, limited gas exchanges, symptoms, and medical history, patients are diagnosed with chronic obstructive pulmonary disease (COPD), a disorder that typically occurs in smokers. However, while COPD usually develops at an advanced age in smokers without AATD, it mostly occurs around the age of 30–50 years old in AATD patients[2][3][4]. Even though smoking has been indicated as the primary trigger for the early development of COPD in individuals with AATD, the disorder can also occur at a younger age in AATD patients who never smoked compared to healthy individuals [5][6]. The mutant protein central to AATD is alpha 1-antitrypsin (AAT), an antiprotease which is primarily functional in the lung [3]. To obtain a comprehensive overview of the disease, the current literature on the pathophysiology of AATD, including AAT and its targets, are summarised. Furthermore, AAT augmentation therapy is discussed, and its controversies considering its effectiveness are addressed. In addition, with the aim of outlining ways to create new strategies for the development of novel assays for measuring the effectiveness of AAT therapies, biomarkers of AAT activity and different protease inhibitors are reviewed.
2. Alpha 1-Antitrypsin and Its Targets—Neutrophil Elastase and Proteinase 3
While the majority of AAT is synthesised in hepatocytes, it is also synthesised (at much lower amounts) in neutrophils, macrophages, intestinal and pulmonary alveolar, airway epithelial cells, and in the cornea[7][8]. The gene encoding AAT is SERPINA1 (14q32.1), which consists of seven exons and six introns. After translation, transcription, and post-translational modifications, AAT is released into the circulation. Via the circulation, it reaches the lungs where it functions as a serine protease inhibitor[9]. In this function, it controls the amount of proteolytic degradation, primarily by targeting the neutrophil-derived serine proteases neutrophil elastase (NE), proteinase 3 (PR3), and cathepsin G (Cath G) [10][11]. The central domain of AAT in the inhibitory process is the reactive centre loop (RCL), an amino acid sequence which is crucial for protease recognition and binding. The AAT-protease docking results in a Michaelis complex, the ‘stressed’ AAT conformation, allowing for proteolysis of the RCL. Cleavage of the RCL flips the protease to the opposite pole of AAT, making it kinetically trapped. Simultaneously, AAT ‘relaxes’ again by entering a stable state (see panels A to C in Figure 1), which can be removed from the circulation.
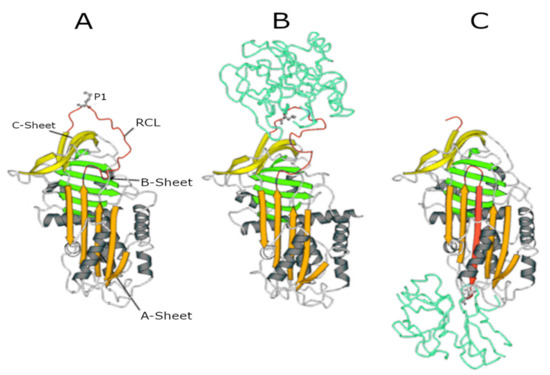
Figure 1. Serpin structures. (A) Native alpha1-antitrypsin (AAT); (B) Michaelis complex between AAT and trypsin; and (C) covalent complex between AAT and trypsin. In all structures, the A-sheet is in orange, the B-sheet is in green, and the C-sheet is in yellow. The reactive centre loop (RCL) in the upper pole of the molecule shows the P1 residue (Met358) recognised by NE.
The fact that AAT is cleaved during this process makes it a single-use inhibition mechanism[7][12][13]. A crucial amino acid in this process is the methionine at position 358 of the enzyme because it is involved in the cleavage of the RCL and the flipping of the protease. This methionine residue, however, is also susceptible to oxidation to methionine sulphoxide. Oxidation, which can result from exposure to endogenously produced reactive oxidants derived from inflammatory cells or exposure to oxidants present in inhaled toxicants such as cigarette smoke, reduces the affinity of AAT for proteases and inactivates AAT[7].
Neutrophil elastase and proteinase 3 are the proteases which are inhibited by AAT and are most important in the development of the AATD phenotype. Cathepsin G, the third proteinase that is cleaved by AAT, does not seem to be involved in this process and will therefore not be considered in this report further [10][11]. Both NE and PR3 are synthesised in neutrophils and stored in an active form in azurophilic granules of the neutrophil [14]. Upon stimulation, neutrophils degranulate and expose the environment to the instantly active proteinases. Upon degranulation, these proteases can be present in a membrane-bound or a soluble form[14][15]. Once released, NE and PR3 are free to cleave their substrates. As these enzymes have 54% of their sequence in common, their substrate specificity is quite similar; both prefer cleaving peptide bonds after small hydrophobic amino acids [16]. Nonetheless, there still are variations in enzyme–substrate interactions, mainly due to differences in the structure of the part of the peptide surrounding the cleavage point (extending beyond P1′; Schechter and Berger nomenclature [17][18].
Although almost all extracellular matrix proteins can be cleaved by NE, clotting factors, complement proteins, immunoglobins, and cytokines are also amongst the targets of this enzyme [19]. When expressed at normal levels, NE predominantly has protective effects in host defence against infection. It aids in the degradation of pathogens by blocking the growth of Gram-negative bacteria[20], fine-tunes tissue remodelling [21], and is involved in the formation of neutrophil extracellular traps [22]. Although NE has beneficial effects when expressed in normal levels, overstimulation of NE production and excessive release cause excessive degradation of extracellular matrix proteins like elastin, collagen, and fibronectin, and cell-associated proteins like E-cadherin. This results in lung parenchyma degradation and disruption of the epithelial barrier due to loss of integrity and shedding of epithelial cells. These processes, when uninhibited, will result in emphysema and chronic inflammation [14][13].
Whereas plenty of literature is available concerning the function of NE, less information is available on the role of PR3. Because PR3 has many of the same targets as NE, it also proteolytically cleaves extracellular matrix proteins. Apart from that, PR3 is involved in early apoptosis[16]. Interestingly, the majority of the literature on PR3 outlines its involvement in GPA, one of the extrapulmonary diseases associated with AATD. GPA is characterised by a high expression of membrane-bound PR3 on neutrophils, which is recognised as an antigen by anti-neutrophil cytoplasmic antibodies (ANCAs) typically present in such patients. In patients with AATD, the lack of AAT causes an increase of active PR3 at the cell surface of neutrophils and thereby increases the risk of developing GPA[22]. This is one example of the important role that AAT has in regulating the balance of proteolytic activity in the lungs. Inhibiting NE and PR3 and thereby preventing unintended inflammation and damage to the lung tissue is a crucial function of AAT, and important for maintaining lung health. Immediately after degranulation, however, NE and PR3 outnumber AAT in the zone close to the cell membrane. In addition, AAT is not able to entirely reach the membrane of the neutrophils, so membrane-bound NE and PR3 will not be inhibited as effectively [23]. This indicates that the control of proteolysis by NE and PR3 is the result of a delicate balance between these proteases and AAT.
An antiprotease which can possibly take on the function of AAT when AAT is absent is alpha2-macroglobulin (α2-M) [11]. As demonstrated in earlier studies, α2-M is capable of irreversible NE inhibition [24]. Although sequestering of NE by α2-M is not very common in healthy individuals because AAT is more effective, it does seem to be important in NE control when AAT is absent[25]. Inhibition of PR3 by α2-M, on the contrary, is poor [26].
References
- Soriano, J.B.; Mahadeva, R. α1-Antitrypsin deficiency: Count me in please! Eur. Respir. J. 2017, 49, 1601941, doi:10.1183/13993003.01941-2016.
- Stockley, R.A.; Turner, A.M. α-1-Antitrypsin deficiency: Clinical variability, assessment, and treatment. Trends Mol. Med. 2014, 20, 105–115, doi:10.1016/j.molmed.2013.11.006.
- McCarthy, C.; Reeves, E.P.; McElvaney, N.G. The Role of Neutrophils in Alpha-1 Antitrypsin Deficiency. Ann. Am. Thorac. Soc. 2016, 13, S297–S304, doi:10.1513/AnnalsATS.201509-634KV.
- Janssen, R.; Piscaer, I.; Franssen, F.M.E.; Wouters, E.F.M. Emphysema: Looking beyond alpha-1 antitrypsin deficiency. Expert Rev. Respir. Med. 2019, 13, 381–397, doi:10.1080/17476348.2019.1580575.
- Alpha-1 foundation: Lung disease alpha1.org. Available online: https://www.alpha1.org/newly-diagnosed/learning-about-alpha-1/lung-disease/ (accessed on 5 June 2020).
- Ferrarotti, I.; Ottaviani, S.; De Silvestri, A.; Corsico, A.G. Update on α(1)-antitrypsin deficiency. Breathe 2018, 14, e17–e24, doi:10.1183/20734735.015018.
- Kalsheker, N. Alpha1-antitrypsin: Structure, function and molecular biology of the gene. Biosci. Rep. 1989, 9, 129–138, doi:10.1007/BF01115992.
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455, doi:10.1056/NEJMra1910234.
- Bashir, A.; Shah, N.N.; Hazari, Y.M.; Habib, M.; Bashir, S.; Hilal, N.; Banday, M.; Asrafuzzaman, S.; Fazili, K.M. Novel variants of SERPIN1A gene: Interplay between alpha1-antitrypsin deficiency and chronic obstructive pulmonary dis-ease. Respir. Med. 2016, 117, 139–149, doi:10.1016/j.rmed.2016.06.005.
- Gooptu, B.; Ekeowa, U.I.; Lomas, D.A. Mechanisms of emphysema in alpha1-antitrypsin deficiency: Molecular and cel-lular insights. Eur. Respir. J. 2009, 34, 475–488, doi:10.1183/09031936.00096508.
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.U.; Dafforn, T.R.; Stockley, R.A. α-1-antitrypsin variants and the protein-ase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L179–L190, doi:10.1152/ajplung.00179.2014.
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Al-pha-1-antitrypsin deficiency: Genetic variations, clinical manifestations and therapeutic interventions. Mutat. Res. 2017, 773, 14–25, doi:10.1016/j.mrrev.2017.03.001.
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001, 276, 33293–33296, doi:10.1074/jbc.R100016200.
- Loke, I.; Østergaard, O.; Heegaard, N.H.H.; Packer, N.H.; Thaysen-Andersen, M. Paucimannose-rich N-glycosylation of spatiotemporally regulated human neutrophil elastase modulates its immune functions. Mol. Cell. Proteom. 2017, 16, 1507–1527, doi:10.1074/mcp.M116.066746.
- Hinkofer, L.C.; Seidel, S.A.; Korkmaz, B.; Silva, F.; Hummel, A.M.; Braun, D.; Jenne, D.E.; Specks, U. A monoclonal anti-body (MCPR3-7) interfering with the activity of proteinase 3 by an allosteric mechanism. J. Biol. Chem. 2013, 288, 26635–26648, doi:10.1074/jbc.M113.495770.
- Martin, K.R.; Witko-Sarsat, V. Proteinase 3: The odd one out that became an autoantigen. J. Leukoc. Biol. 2017, 102, 689–698, doi:10.1189/jlb.3MR0217-069R.
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162, doi:10.1016/s0006-291x(67)80055-x.
- Hajjar, E.; Korkmaz, B.; Reuter, N. Differences in the substrate binding sites of murine and human proteinase 3 and neu-trophil elastase. FEBS Lett. 2007, 581, 5685–5690, doi:10.1016/j.febslet.2007.11.029.
- Lee, W.L.; Downey, G.P. Leukocyte elastase: Physiological functions and role in acute lung injury. Am. J. Respir. Crit. Care Med. 2001, 164, 896–904, doi:10.1164/ajrccm.164.5.2103040.
- Belaaouaj, A. Neutrophil elastase-mediated killing of bacteria: Lessons from targeted mutagenesis. Microbes Infect. 2002, 4, 1259–1264, doi:10.1016/s1286-4579(02)01654-4.
- Lungarella, G.; Cavarra, E.; Lucattelli, M.; Martorana, P.A. The dual role of neutrophil elastase in lung destruction and repair. Int. J. Biochem. Cell Biol. 2008, 40, 1287–1296, doi:10.1016/j.biocel.2007.12.008.
- Mota, A.; Sahebghadam Lotfi, A.; Jamshidi, A.R.; Najavand, S. Alpha 1-antitrypsin activity is markedly decreased in Wegener’s granulomatosis. Rheumatol. Int. 2014, 34, 553–558, doi:10.1007/s00296-013-2745-9.
- Von Nussbaum, F.; Li, V.M.J. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clini-cal testing with pre-adaptive pharmacophores. Bioorg. Med. Chem. Lett. 2015, 25, 4370–4381, doi:10.1016/j.bmcl.2015.08.049.
- Salvesen, G.; Virca, G.D.; Travis, J. Interaction of alpha 2-macroglobulin with neutrophil and plasma proteinases. Ann. N. Y. Acad. Sci. 1983, 421, 316–326, doi:10.1111/j.1749-6632.1983.tb18120.x.
- Virca, G.D.; Travis, J. Kinetics of association of human proteinases with human alpha 2-macroglobulin. J. Biol. Chem. 1984, 259, 8870–8874.
- N’Guessan, K.; Grzywa, R.; Seren, S.; Gabant, G.; Juliano, M.A.; Moniatte, M.; van Dorsselaer, A.; Bieth, J.G.; Kellen-berger, C.; Gauthier, F.; et al. Human proteinase 3 resistance to inhibition extends to alpha-2 macroglobulin. FEBS J. 2020, doi:10.1111/febs.15229.
- Virca, G.D.; Travis, J. Kinetics of association of human proteinases with human alpha 2-macroglobulin. J. Biol. Chem. 1984, 259, 8870–8874.
- N’Guessan, K.; Grzywa, R.; Seren, S.; Gabant, G.; Juliano, M.A.; Moniatte, M.; van Dorsselaer, A.; Bieth, J.G.; Kellen-berger, C.; Gauthier, F.; et al. Human proteinase 3 resistance to inhibition extends to alpha-2 macroglobulin. FEBS J. 2020, doi:10.1111/febs.15229.

