2. Alpha 1-Antitrypsin and Its Targets—Neutrophil Elastase and Proteinase 3
While the majority of AAT is synthesised in hepatocytes, it is also synthesised (at much lower amounts) in neutrophils, macrophages, intestinal and pulmonary alveolar, airway epithelial cells, and in the cornea [
7,
8]. The gene encoding AAT is
SERPINA1 (
14q32.1), which consists of seven exons and six introns. After translation, transcription, and post-translational modifications, AAT is released into the circulation. Via the circulation, it reaches the lungs where it functions as a serine protease inhibitor [
9]. In this function, it controls the amount of proteolytic degradation, primarily by targeting the neutrophil-derived serine proteases neutrophil elastase (NE), proteinase 3 (PR3), and cathepsin G (Cath G) [
10,
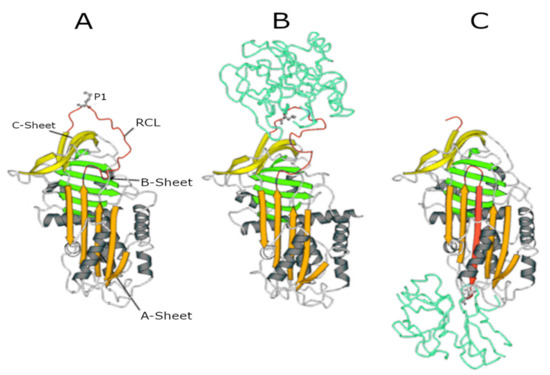
11]. The central domain of AAT in the inhibitory process is the reactive centre loop (RCL), an amino acid sequence which is crucial for protease recognition and binding. The AAT-protease docking results in a Michaelis complex, the ‘stressed’ AAT conformation, allowing for proteolysis of the RCL. Cleavage of the RCL flips the protease to the opposite pole of AAT, making it kinetically trapped. Simultaneously, AAT ‘relaxes’ again by entering a stable state (see panels A to C in ), which can be removed from the circulation.
Figure 1. Serpin structures. (A) Native alpha1-antitrypsin (AAT); (B) Michaelis complex between AAT and trypsin; and (C) covalent complex between AAT and trypsin. In all structures, the A-sheet is in orange, the B-sheet is in green, and the C-sheet is in yellow. The reactive centre loop (RCL) in the upper pole of the molecule shows the P1 residue (Met358) recognised by NE.
The fact that AAT is cleaved during this process makes it a single-use inhibition mechanism [
7,
12,
13]. A crucial amino acid in this process is the methionine at position 358 of the enzyme because it is involved in the cleavage of the RCL and the flipping of the protease. This methionine residue, however, is also susceptible to oxidation to methionine sulphoxide. Oxidation, which can result from exposure to endogenously produced reactive oxidants derived from inflammatory cells or exposure to oxidants present in inhaled toxicants such as cigarette smoke, reduces the affinity of AAT for proteases and inactivates AAT [
7].
Neutrophil elastase and proteinase 3 are the proteases which are inhibited by AAT and are most important in the development of the AATD phenotype. Cathepsin G, the third proteinase that is cleaved by AAT, does not seem to be involved in this process and will therefore not be considered in this report further [
10,
11]. Both NE and PR3 are synthesised in neutrophils and stored in an active form in azurophilic granules of the neutrophil [
14]. Upon stimulation, neutrophils degranulate and expose the environment to the instantly active proteinases. Upon degranulation, these proteases can be present in a membrane-bound or a soluble form [
14,
15]. Once released, NE and PR3 are free to cleave their substrates. As these enzymes have 54% of their sequence in common, their substrate specificity is quite similar; both prefer cleaving peptide bonds after small hydrophobic amino acids [
16]. Nonetheless, there still are variations in enzyme–substrate interactions, mainly due to differences in the structure of the part of the peptide surrounding the cleavage point (extending beyond P1′; Schechter and Berger nomenclature [
17]) [
18].
Although almost all extracellular matrix proteins can be cleaved by NE, clotting factors, complement proteins, immunoglobins, and cytokines are also amongst the targets of this enzyme [
19]. When expressed at normal levels, NE predominantly has protective effects in host defence against infection. It aids in the degradation of pathogens by blocking the growth of Gram-negative bacteria [
20], fine-tunes tissue remodelling [
21], and is involved in the formation of neutrophil extracellular traps [
22]. Although NE has beneficial effects when expressed in normal levels, overstimulation of NE production and excessive release cause excessive degradation of extracellular matrix proteins like elastin, collagen, and fibronectin, and cell-associated proteins like E-cadherin. This results in lung parenchyma degradation and disruption of the epithelial barrier due to loss of integrity and shedding of epithelial cells. These processes, when uninhibited, will result in emphysema and chronic inflammation [
14,
23].
Whereas plenty of literature is available concerning the function of NE, less information is available on the role of PR3. Because PR3 has many of the same targets as NE, it also proteolytically cleaves extracellular matrix proteins. Apart from that, PR3 is involved in early apoptosis [
16]. Interestingly, the majority of the literature on PR3 outlines its involvement in GPA, one of the extrapulmonary diseases associated with AATD. GPA is characterised by a high expression of membrane-bound PR3 on neutrophils, which is recognised as an antigen by anti-neutrophil cytoplasmic antibodies (ANCAs) typically present in such patients. In patients with AATD, the lack of AAT causes an increase of active PR3 at the cell surface of neutrophils and thereby increases the risk of developing GPA [
24]. This is one example of the important role that AAT has in regulating the balance of proteolytic activity in the lungs. Inhibiting NE and PR3 and thereby preventing unintended inflammation and damage to the lung tissue is a crucial function of AAT, and important for maintaining lung health. Immediately after degranulation, however, NE and PR3 outnumber AAT in the zone close to the cell membrane. In addition, AAT is not able to entirely reach the membrane of the neutrophils, so membrane-bound NE and PR3 will not be inhibited as effectively [
25]. This indicates that the control of proteolysis by NE and PR3 is the result of a delicate balance between these proteases and AAT.
An antiprotease which can possibly take on the function of AAT when AAT is absent is alpha2-macroglobulin (α2-M) [
11]. As demonstrated in earlier studies, α2-M is capable of irreversible NE inhibition [
26]. Although sequestering of NE by α2-M is not very common in healthy individuals because AAT is more effective, it does seem to be important in NE control when AAT is absent [
27]. Inhibition of PR3 by α2-M, on the contrary, is poor [
28].