 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kay Ohlendieck | + 3918 word(s) | 3918 | 2021-02-16 04:38:44 | | | |
| 2 | Dean Liu | -1864 word(s) | 2054 | 2021-02-19 09:08:44 | | |
Video Upload Options
Dystrophin isoform Dp427-M is tightly associated with a variety of glycoproteins at the muscle sarcolemma membrane. The core dystrophin-glycoprotein complex forms a variety of links to components of the extracellular matrix and the intracellular cytoskeleton. The wider dystrophin complexome plays a crucial functional role as an integrative node of the skeletal muscle periphery. The sarcolemmal dystrophin node is involved in the maintenance of fiber stability, the provision of cellular signaling cascades, organizer of cytoskeletal networks and costameric anchor for lateral force transmission.
1.The Core Dystrophin Complex in Skeletal Muscle
The full-length dystrophin isoform Dp427-M belongs to the class of giant muscle proteins[1] and consists of several distinct molecular domains as illustrated in the upper panel of Figure 1. This includes amino-terminal and central actin-binding domains, proline-rich hinge regions, spectrin-like rod domains and crucial carboxy-terminal binding sites for interactions with plasmalemmal and cytosolic components[2][3][4][5]. Dystrophin closely interacts with the integral proteins beta-dystroglycan, alpha/beta/gamma/delta-sarcoglyan and sarcospan of the sarcolemma, the extracellular receptor alpha-dystroglycan and laminin-211, the cytosolic components alpha/beta-dystrobrevin and alpha/beta-syntrophin, and the cortical actin cytoskeleton[6][7][8][9][10], as shown in the lower panel of Figure 1.
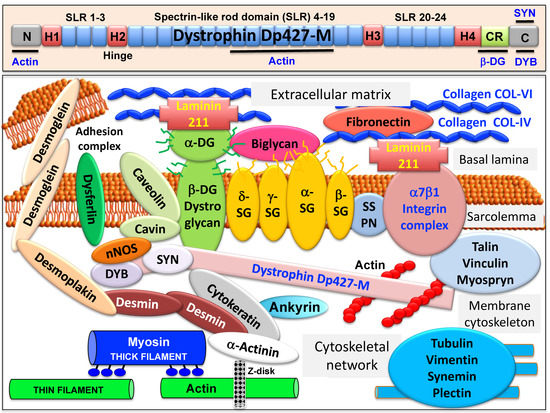
Figure 1. Overview of the domain structure of dystrophin and the diverse interactions of the dystrophin–glycoprotein complex in skeletal muscle tissues. The upper panel shows a diagrammatic presentation of the main molecular domains of dystrophin isoform Dp427-M, including actin-binding sites at the N-terminus and central rod domain, proline-rich hinge regions (H1 to H4), spectrin-like rod (SLR) domains 1–3, 4–19 and 20–24, a cysteine-rich domain with binding sites for integral beta-dystroglycan (DG), the cysteine-rich domain (CR) and the C-terminus with binding sites for dystrobrevin (DYB) and syntrophin (SYN). The lower panel shows a model of the spatial configuration of the dystrophin complexome in skeletal muscle fibers. Shown is the dystrophin core complex consisting of the dystrophin isoform Dp427-M, dystroglycans (DG), sarcoglycans (SG), sarcospan (SSPN), syntrophins (SYN) and dystrobrevins (DYB), as well as the wider dystrophin-associated network that forms associations with the extracellular matrix, the sarcolemma, the cytoskeleton and the sarcomere.
Sedimentation analysis of the isolated dystrophin complex suggests a monomeric structure with an apparent molecular mass of 1.2 MDa[11]. In Duchenne muscular dystrophy, alterations in the expression of members of the dystrophin network are closely related to key pathophysiological features in dystrophin-deficient muscles, including degeneration-regeneration cycles, reactive myo-fibrosis, fat substitution and sterile inflammation. Detailed reviews have been published on the composition of the core dystrophin complex[12][13][14][15], as well as the role of dystrophin and its associated glycoprotein complex in the multisystemic complications of dystrophinopathy and pathophysiological crosstalk throughout the body[16][17][18][19][20]. Here we refer to specific aspects of dystrophin interactions that are crucial for our general understanding of the wider functional role of the dystrophin complexome in normal skeletal muscle tissue.
2. The Dystrophin Node in Skeletal Muscle
A model of the spatial configuration of the core dystrophin complex and its association with the extracellular matrix on the one hand and the intracellular cytoskeletal network of contractile fibers on the other hand is presented in the lower panel of above Figure 1. The cell biological concept that the dystrophin–glycoprotein complex occupies a central position at the fiber periphery is summarized in Figure 2. The diagram shows that the dystrophin-associated surface complex forms an organizing node that is majorly involved in (i) the provision of sarcolemmal membrane integrity via a stabilizing linkage between the intracellular actin cytoskeleton and the extracellular matrix protein laminin[8][10], (ii) the establishment of a molecular scaffold and anchoring system for ion channels and enzymes to mediate cellular signaling processes[21][22] (iii) the organization of actin filament attachment and its associated cytoskeletal network[23][24], and (iv) the mediation of lateral force transmission from sarcomeric contraction to the endomysium and its connected layers of the extracellular matrix[25][26].
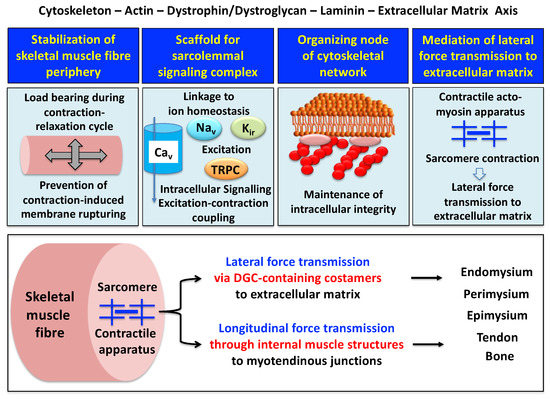
Figure 2. Outline of the main functions of the dystrophin node and its associated protein complex as integrators of fiber stability, cellular signaling, cytoskeletal organization and lateral force transmission. The upper panels summarize the main functions of the trans-sarcolemmal axis formed by the intracellular actin cytoskeleton, the dystrophin–dystroglycan complex, the basal lamina component laminin and the extracellular matrix. The lower panel illustrates the physiological concept of force transmission in skeletal muscles, which can be divided into a laterally and a longitudinally acting system. In conjunction with other costameric proteins, the dystrophin–glycoprotein complex (DGC) is majorly involved in lateral force transmission to the extracellular matrix.
3. The Sarcolemmal Dystrophin Complex and Lateral Force Transmission
The peripheral structure of skeletal muscle fibers functions as an essential physical barrier with its protective basal lamina. The underlying sarcolemma membrane provides the physiological structure for the efficient exchange of ions, metabolites and signaling molecules within the contractile system[27]. The plasmalemma is connected to the terminal cisternae region of the sarcoplasmic reticulum at the triad junctions via its invaginations, the transverse tubules. This intricate membrane assembly and its associated Ca2+-handling apparatus is involved in the fine regulation of excitation–contraction coupling, muscle relaxation and ion homeostasis, and encounters enormous physical strain during contraction–relaxation cycles[28]. The dystrophin-associated complex is implicated to act as a biomolecular shock absorber by linking the basal lamina to the actin cytoskeleton and thus preventing rupturing of this muscle membrane system[29][30][31].
At specialized costamere regions, which play both a mechanical and a signaling role, the dystrophin complex forms in conjunction with the integrin–vinculin–talin axis a link to the contractile sarcomere units[24][32]. This bridging structure is postulated to provide an indirect means of lateral force transmission to the collagen-rich muscle exterior, in addition to longitudinal forces that are transmitted directly from the contractile apparatus through the cytosol to the myotendinous junction[33][34][35]. In skeletal muscle fibers, the characteristic longitudinal pattern of A bands and I bands reflect the organization of myosin-containing thick filaments and actin-containing thin filaments with their contractile sarcomeric units, which are positioned between Z discs. Following the energy-dependent crossbridge coupling between myosin heads and actin filaments, thin filaments slide past thick filaments. The force generated by this sarcomeric shortening event is partially transmitted by a lateral direct force between Z-disk structures and the M-line regions of neighboring myofibrils. Costamere structures at the fiber periphery play a central role as sensors of the relative mechanical load and support force transduction across the muscle plasma membrane. Contractile force is then further transmitted to the complex layers of the extracellular matrix, consisting of endomysium, perimysium and epimysium, towards the tendon and bone structure[26][33][34]. The second type of force transmission mechanism works by longitudinal means through internal muscle structures embedded in the cytosol. Both lateral and longitudinal coupling mechanisms act in parallel and ultimately transmit the force generated by the actomyosin apparatus in the sarcomere to the tendon and anchoring structures such as bone, as diagrammatically summarized in the lower panel of Figure 2. The dystrophin-associated dystroglycan subcomplex was shown to play a critical role in the sarcomeric cytoskeleton by limiting contraction-induced injury to skeletal muscle fibers[31].
The elucidation of the multifaceted functions of the dystrophin–glycoprotein complex in maintaining membrane stability during excitation–contraction–relaxation cycles, assisting lateral force transmission through costameres and providing a scaffold for anchoring surface receptors and maintaining cellular signaling mechanisms was carried out by multidisciplinary approaches. This included molecular genetics, biochemical purification strategies, structural/biophysical analysis, mass spectrometric proteomics analysis, bioinformatics, chemical crosslinking, cell biological characterization and comparative biomedical studies.
4. Muscle Dystrophin Dp427-M and Its Associated Glycoprotein Complex
The large muscle isoform of dystrophin is a rod-shaped protein[36] with considerable homology to the actinin superfamily of actin crosslinking components, which also includes utrophin and spectrin[37]. Both, dystrophin isoform Dp427-M of the sarcolemma and its autosomal homologue, utrophin isoform Up395-M of the neuromuscular junction, exhibit typical biochemical properties of cytoskeletal components, such as insolubility in non-ionic detergent and efficient extraction by alkaline treatment[38][39]. Compared to the main components of the contractile actomyosin apparatus and its regulatory sarcomeric elements, dystrophin represents a relatively minor component of the skeletal muscle fiber proteome. However, dystrophin isoform Dp427-M constitutes a considerable fraction of the subsarcolemmal cytoskeleton in contractile tissue[38]. This makes full-length dystrophin an important structural and functional component of the sarcolemmal lattice and costamere structures [32]. Besides being present in contractile fibers, dystrophin isoforms also exist in many non-muscle cells[40]. The various dystrophins are encoded by the 79 exon-spanning DMD gene, whereby seven different promoters drive the tissue-specific expression of the full-length isoforms Dp427-B in brain, Dp427-M in muscle and Dp427-P in Purkinje cells[41], as well as the shorter isoforms Dp260-R in retina[42], Dp140-B/K in brain and kidney[43], Dp116-S in Schwann cells[44] and Dp71-G in the brain[45] and a variety of other tissues including the spleen[46]. The promoter for Dp71 also produces the shortest known dystrophin isoform named Dp45, which is located in the central nervous system[47]. Of note, the central nervous system displays one of the greatest varieties of dystrophin isoforms, which are involved in synaptic modulation, neuronal excitability and signal integration. Brain Dp427-B is present in neurons of the cerebral cortex and in cerebellar Purkinje cells, Dp140-B is highly expressed during brain development and Dp71-G is located in both neurons and glia cells in the dentate gyrus[45]. Cognitive impairments and emotional disturbances in Duchenne patients are probably linked to altered dystrophin expression in the central nervous system and this is reflected by structural brain abnormalities[48]. The formation of dystrophin complexes and their involvement in dystrophinopathy-associated brain defects has been reviewed by Waite et al.[49].
The composition of the dystrophin–glycoprotein complex has been extensively investigated using a combination of digitonin-based solubilization, wheat germ agglutinin lectin chromatography, ion exchange chromatography and density gradient ultracentrifugation[50][51][11][52], as well as various chemical crosslinking and immunoprecipitation approaches[53][54][55][56][57][58]. Differential detergent extraction procedures[59], two-dimensional gel electrophoresis[7] or alkaline dissociation[52] can be used to isolate individual dystrophin subcomplexes or separate the dystrophin-associated glycoprotein complex from homogeneous dystrophin molecules. Based on these analyses, the core members of the dystrophin-associated complex can be divided into (i) cytosolic components alpha/beta-dystrobrevin [60][61] and alpha/beta-syntrophin[62][63] that interact with the cysteine-rich domain of dystrophin; (ii) integral glycoproteins, including the alpha/beta/gamma/delta-sarcoglyan subcomplex[64][65][66], the highly hydrophobic protein sarcospan[67][68][69] and the main carboxy-terminal dystrophin-binding partner beta-dystroglycan[70]; (iii) laminin-211 [71] and its extracellular receptor alpha-dystroglycan[72], which is a proteolytic cleavage product of the pre-dystroglycan molecule[73]; and (iv) the intracellular actin cytoskeleton that links to an amino-terminal and a rod domain site of full-length dystrophin[2][11][74].
The sarcoglycan subcomplex is initially assembled by the formation of a core between beta-sarcoglycan and delta-sarcoglycan, which subsequently recruits the other two sarcoglycans[66]. Through interactions with sarcospan and additional dystrophin-associated proteins, the sarcoglycan complex secures the formation and mechanical maintenance of the sarcolemmal dystrophin complex. Besides its integrating role in membrane stabilization, the sarcoglycan subcomplex can be chemically modified during fiber contraction, which provides the transduction of information on relative contractile force into cellular signaling[66]. Interestingly, both components of the dystroglycan subcomplex are products of the same gene, DAG1, which encodes a pre-pro-protein version of alpha/beta-dystroglycan that includes a signaling peptide and both subunits[70][75]. The precursor protein is extensively modified by N- and O-glycosylation and undergoes proteolytic processing that generates the integral glycoprotein beta-dystroglycan and the extracellular laminin-binding receptor alpha-dystroglycan[72]. Thus, the two dystroglycans form the backbone of the trans-sarcolemmal linkage between the basal lamina component laminin-211 and the dystrophin-associated actin cytoskeleton in the subsarcolemmal region of skeletal muscle[8][10]. The phosphorylation of beta-dystroglycan, especially intracellular tyrosine residues[76], is a crucial step during interactions with signaling proteins[77]. The phosphorylation of the cysteine-rich region in the carboxy-terminal domain of dystrophin also plays a key role in strengthening the interaction with beta-dystroglycan. Thus, post-translational modifications are important modulators of dynamic associations within the dystrophin–dystroglycan axis.
5. The Dystrophin Complex as a Cellular Signaling Node in Skeletal Muscle
Besides providing the above-described stabilizing linkage between the basal lamina and the membrane cytoskeleton and thereby functioning as a molecular shock absorber, the dystrophin complex also acts as a critical hub for cellular signaling at the muscle plasma membrane[22]. The dystrophin complexome has been implicated to be involved in the modulation of hypertrophy, major kinase signaling cascades, the organization of caveolae structures, the regulation of skeletal muscle size, the mitogen-activated protein kinase pathway, the regulation of ion homeostasis, cytoskeletal organization, G-protein signaling and neuromuscular transmission in conjunction with its autosomal homologue utrophin, as well as mechano-sensing and cytoskeletal remodeling in association with the laminin-collagen bridge and the sarcolemmal integrin complex[18][22][78].
References
- Koenig, M.; Kunkel, L.M. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 1990, 265, 4560–4566.
- Fealey, M.E.; Horn, B.; Coffman, C.; Miller, R.; Lin, A.Y.; Thompson, A.R.; Schramel, J.; Groth, E.; Hinderliter, A.; Cembran, A.; et al. Dynamics of dystrophin’s actin-binding domain. Biophys. J. 2018, 115, 445–454.
- Abdullah, M.; Hassan, A.; Rashid, S.; Naeem, M. Tyrosine phosphorylation as a regulator of dystrophin and beta-dystroglycan interaction: A molecular insight. J. Mol. Graph Model. 2020, 99, 107623.
- Delalande, O.; Molza, A.E.; Dos Santos Morais, R.; Chéron, A.; Pollet, É.; Raguenes-Nicol, C.; Tascon, C.; Giudice, E.; Guilbaud, M.; Nicolas, A.; et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J. Biol. Chem. 2018, 293, 6637–6646.
- Rybakova, I.N.; Ervasti, J.M. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J. Biol. Chem. 1997, 272, 28771–28778.
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131.
- Yamamoto, H.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Ozawa, E. Heterogeneity of dystrophin-associated proteins. J. Biochem. 1993, 114, 132–139. sti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823.
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679.
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10.
- Murphy, S.; Ohlendieck, K. Mass spectrometric identification of dystrophin, the protein product of the Duchenne muscular dystrophy gene, in distinct muscle surface membranes. Int. J. Mol. Med. 2017, 40, 1078–1088.
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239.
- Lewis, C.; Ohlendieck, K. Mass spectrometric identification of dystrophin isoform Dp427 by on-membrane digestion of sarcolemma from skeletal muscle. Anal. Biochem. 2010, 404, 197–203.
- Gawor, M.; Prószyński, T.J. The molecular cross talk of the dystrophin-glycoprotein complex. Ann. NY Acad. Sci. 2018, 1412, 62–72.
- Bhat, H.F.; Mir, S.S.; Dar, K.B.; Bhat, Z.F.; Shah, R.A.; Ganai, N.A. ABC of multifaceted dystrophin glycoprotein complex (DGC). J. Cell. Physiol. 2018, 233, 5142–5159.
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697.
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy associated myofibrosis. Proteomics 2016, 16, 345–366.
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305.
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 2018, 8, 1313–1356.
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018, 58, 167–177.
- Leyva-Leyva, M.; Sandoval, A.; Felix, R.; González-Ramírez, R. Biochemical and functional interplay between Ion channels and the components of the dystrophin-associated glycoprotein complex. J. Membr. Biol. 2018, 251, 535–550.
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642.
- Rybakova, I.N.; Amann, K.J.; Ervasti, J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996, 135, 661–672.
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214.
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88.
- Ramaswamy, K.S.; Palmer, M.L.; van der Meulen, J.H.; Renoux, A.; Kostrominova, T.Y.; Michele, D.E.; Faulkner, J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011, 589, 1195–1208.
- Campbell, K.P.; Stull, J.T. Skeletal muscle basement membrane-sarcolemma-cytoskeleton interaction minireview series. J. Biol. Chem. 2003, 278, 12599–12600.
- Shishmarev, D. Excitation-contraction coupling in skeletal muscle: Recent progress and unanswered questions. Biophys. Rev. 2020, 12, 143–153.
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714.
- Sarkis, J.; Vié, V.; Winder, S.J.; Renault, A.; Le Rumeur, E.; Hubert, J.F. Resisting sarcolemmal rupture: Dystrophin repeats increase membrane-actin stiffness. FASEB J. 2013, 27, 359–367.
- Rader, E.P.; Turk, R.; Willer, T.; Beltrán, D.; Inamori, K.; Peterson, T.A.; Engle, J.; Prouty, S.; Matsumura, K.; Saito, F.; et al. Role of dystroglycan in limiting contraction-induced injury to the sarcomeric cytoskeleton of mature skeletal muscle. Proc. Natl. Acad. Sci. USA 2016, 113, 10992–10997.
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594.
- Hughes, D.C.; Wallace, M.A.; Baar, K. Effects of aging, exercise, and disease on force transfer in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1–E10.
- Jaka, O.; Casas-Fraile, L.; López de Munain, A.; Sáenz, A. Costamere proteins and their involvement in myopathic processes. Expert Rev. Mol. Med. 2015, 17, e12.
- Purslow, P.P. The Structure and Role of Intramuscular Connective Tissue in Muscle Function. Front. Physiol. 2020, 11, 495.
- Murphy, A.C.; Young, P.W. The actinin family of actin cross-linking proteins—A genetic perspective. Cell. Biosci. 2015, 5, 49.
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228.
- Ohlendieck, K.; Campbell, K.P. Dystrophin constitutes 5% of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991, 283, 230–234.
- Ohlendieck, K. Characterisation of the dystrophin-related protein utrophin in highly purified skeletal muscle sarcolemma vesicles. Biochim. Biophys. Acta 1996, 1283, 215–222.
- Tokarz, S.A.; Duncan, N.M.; Rash, S.M.; Sadeghi, A.; Dewan, A.K.; Pillers, D.A. Redefinition of dystrophin isoform distribution in mouse tissue by RT-PCR implies role in nonmuscle manifestations of duchenne muscular dystrophy. Mol. Genet. Metab. 1998, 65, 272–281.
- Ueda, H.; Baba, T.; Ohno, S. Current knowledge of dystrophin and dystrophin-associated proteins in the retina. Histol. Histopathol. 2000, 15, 753–760.
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740.
- Lidov, H.G.; Kunkel, L.M. Dystrophin and Dp140 in the adult rodent kidney. Lab. Invest. 1998, 78, 1543–1551.
- Mahyoob Rani, A.Q.; Maeta, K.; Kawaguchi, T.; Awano, H.; Nagai, M.; Nishio, H.; Matsuo, M. Schwann cell-specific Dp116 is expressed in glioblastoma cells, revealing two novel DMD gene splicing patterns. Biochem. Biophys. Rep. 2019, 20, 100703.
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the neuropathophysiology of duchenne muscular dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767.
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide changes in the mdx-4cv spleen due to pathophysiological crosstalk with dystrophin-deficient skeletal muscle. iScience 2020, 23, 101500.
- Romo-Yáñez, J.; Rodríguez-Martínez, G.; Aragón, J.; Siqueiros-Márquez, L.; Herrera-Salazar, A.; Velasco, I.; Montanez, C. Characterization of the expression of dystrophins and dystrophin-associated proteins during embryonic neural stem/progenitor cell differentiation. Neurosci. Lett. 2020, 736, 135247.
- Angelini, C.; Pinzan, E. Advances in imaging of brain abnormalities in neuromuscular disease. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419845567.
- Waite, A.; Brown, S.C.; Blake, D.J. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 2012, 35, 487–496.
- Ervasti, J.M.; Kahl, S.D.; Campbell, K.P. Purification of dystrophin from skeletal muscle. J. Biol. Chem. 1991, 266, 9161–9165.
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319.
- Ohlendieck, K.; Ervasti, J.M.; Snook, J.B.; Campbell, K.P. Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J. Cell Biol. 1991, 112, 135–148.
- Finn, D.M.; Ohlendieck, K. Oligomerization of beta-dystroglycan in rabbit diaphragm and brain as revealed by chemical crosslinking. Biochim. Biophys. Acta 1998, 1370, 325–336.
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752.
- Johnson, E.K.; Li, B.; Yoon, J.H.; Flanigan, K.M.; Martin, P.T.; Ervasti, J.; Montanaro, F. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE 2013, 8, e73224.
- Turk, R.; Hsiao, J.J.; Smits, M.M.; Ng, B.H.; Pospisil, T.C.; Jones, K.S.; Campbell, K.P.; Wright, M.E. Molecular signatures of membrane protein complexes underlying muscular dystrophy. Mol. Cell. Proteom. 2016, 15, 2169–2185.
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell. Motil. 2017, 38, 251–268.
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17.
- Yoshida, M.; Suzuki, A.; Yamamoto, H.; Noguchi, S.; Mizuno, Y.; Ozawa, E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur. J. Biochem. 1994, 222, 1055–1061.
- Sadoulet-Puccio, H.M.; Feener, C.A.; Schaid, D.J.; Thibodeau, S.N.; Michels, V.V.; Kunkel, L.M. The genomic organization of human dystrobrevin. Neurogenetics 1997, 1, 37–42.
- Nakamori, M.; Takahashi, M.P. The role of alpha-dystrobrevin in striated muscle. Int. J. Mol. Sci. 2011, 12, 1660–1671.
- Gee, S.H.; Gee, R.; Madhaven, S.H.; Levinson, S.R.; Caldwell, J.H.; Sealock, R.; Froehner, S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 1998, 18, 128–137.
- Bhat, S.S.; Ali, R.; Khanday, F.A. Syntrophins entangled in cytoskeletal meshwork: Helping to hold it all together. Cell Prolif. 2019, 52, e12562.
- Roberds, S.L.; Anderson, R.D.; Ibraghimov-Beskrovnaya, O.; Campbell, K.P. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin). J. Biol. Chem. 1993, 268, 23739–23742.
- Anastasi, G.; Cutroneo, G.; Rizzo, G.; Favaloro, A. Sarcoglycan subcomplex in normal and pathological human muscle fibers. Eur. J. Histochem. 2007, 51, 29–33.
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci. 2016, 21, 744–756.
- Crosbie, R.H.; Heighway, J.; Venzke, D.P.; Lee, J.C.; Campbell, K.P. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J. Biol. Chem. 1997, 272, 31221.
- Crosbie, R.H.; Lim, L.E.; Moore, S.A.; Hirano, M.; Hays, A.P.; Maybaum, S.W.; Collin, H.; Dovico, S.A.; Stolle, C.A.; Fardeau, M.; et al. Molecular and genetic characterization of sarcospan: Insights into sarcoglycan-sarcospan interactions. Hum. Mol. Genet. 2000, 9, 2019–2027.
- Marshall, J.L.; Crosbie-Watson, R.H. Sarcospan: A small protein with large potential for Duchenne muscular dystrophy. Skelet. Muscle 2013, 3, 1.
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702.
- Carmignac, V.; Durbeej, M. Cell-matrix interactions in muscle disease. J. Pathol. 2012, 226, 200–218.
- Bozzi, M.; Morlacchi, S.; Bigotti, M.G.; Sciandra, F.; Brancaccio, A. Functional diversity of dystroglycan. Matrix Biol. 2009, 28, 179–187.
- Barresi, R.; Campbell, K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006, 119, 199–207.
- Mias-Lucquin, D.; Dos Santos Morais, R.; Chéron, A.; Lagarrigue, M.; Winder, S.J.; Chenuel, T.; Pérez, J.; Appavou, M.S.; Martel, A.; Alviset, G.; et al. How the central domain of dystrophin acts to bridge F-actin to sarcolemmal lipids. J. Struct. Biol. 2020, 209, 107411.
- Ibraghimov-Beskrovnaya, O.; Milatovich, A.; Ozcelik, T.; Yang, B.; Koepnick, K.; Francke, U.; Campbell, K.P. Human dystroglycan: Skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum. Mol. Genet. 1993, 2, 1651–1657.
- Miller, G.; Moore, C.J.; Terry, R.; La Riviere, T.; Mitchell, A.; Piggott, R.; Dear, T.N.; Wells, D.J.; Winder, S.J. Preventing phosphorylation of dystroglycan ameliorates the dystrophic phenotype in mdx mouse. Hum. Mol. Genet. 2012, 21, 4508–4520.
- Moore, C.J.; Winder, S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul. Disord. 2012, 22, 959–965.
- Swiderski, K.; Shaffer, S.A.; Gallis, B.; Odom, G.L.; Arnett, A.L.; Scott Edgar, J.; Baum, D.M.; Chee, A.; Naim, T.; Gregorevic, P.; et al. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum. Mol. Genet. 2014, 23, 6697–6711.
- Jung, D.; Yang, B.; Meyer, J.; Chamberlain, J.S.; Campbell, K.P. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J. Biol. Chem. 1995, 270, 27305–27310.

