 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mathijs Scholtes | + 2186 word(s) | 2186 | 2021-02-08 02:03:49 | | | |
| 2 | Peter Tang | -2 word(s) | 2184 | 2021-02-17 13:03:49 | | |
Video Upload Options
Metabolic reprogramming (MR) is an upregulation of biosynthetic and bioenergetic pathways to satisfy increased energy and metabolic building block demands of tumors. This includes glycolytic activity, which deprives the tumor microenvironment (TME) of nutrients while increasing extracellular lactic acid.
1. Introduction
Worldwide, there were approximately 550,000 new cases and 200,000 deaths from bladder cancer (BC) in 2018 [1]. Up to 90% of BC cases originate from the luminal urothelial lining of the bladder and produce urothelial carcinomas (UC). Non-muscle invasive UC (NMIUC) accounts for 75% of BC patients and is commonly treated by transurethral tumor resection (TUR) with and without adjuvant intravesical instillations [2]. In contrast, muscle-invasive UC (MIUC) is treated with either cisplatin-based neoadjuvant chemotherapy (NAC) followed by surgical removal of the bladder (cystectomy) or external beam radiotherapy with or without chemotherapy. Despite extensive treatment, half of MIUC patients will progress to metastatic urothelial carcinoma (mUC) [3]. First-line treatment option in mUC is gemcitabine + cisplatin ("gem/cis") [3][4]. However, 30% of mUC patients are cisplatin-ineligible due to poor performance status and other comorbidities [5][6]. Cisplatin-ineligible patients, mostly due to renal compromise, are treated with gemcitabine + carboplatin ("gem/carbo"), which is less effective than cisplatin combinations [7]. Regardless of the platinum-based chemotherapy used, most mUC patients will ultimately progress [8]. In recent years, immune checkpoint therapy (ICT) has emerged as a new option for platinum-relapsed or cisplatin-ineligible patients [3]. ICT targets cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed death (ligand) 1 (PD-1/PDL1), used by tumor cells to inhibit anticancer immune responses [9]. ICT has shown superior efficacy over 2nd line chemotherapy in platinum-relapsed mUC patients [10][11][12]. Currently, several PD-1/PD-L1 inhibitors have been FDA, and EMA approved for the treatment of mUC in the first line (no prior platinum-based chemotherapy) and/or second-line (after the failure of platinum-based chemotherapy) [10][11][12][13][14]. Approved agents used PD-L1 inhibitors: atezolizumab, durvalumab, and avelumab, and PD-1 inhibitors: nivolumab and pembrolizumab [10][11][12]. Treatment of platinum-relapsed mUC patients with pembrolizumab, nivolumab, or atezolizumab was associated with an ORR of ~20% [10][11][12]. Robust biomarkers that can predict clinical response to ICT are lacking due to the complexity of tumor-immune interactions that contribute to ICT resistance [15]. However, it was found recently that a mechanism associated with resistance to ICT is metabolic competition between immune cells and cancer cells in the tumor microenvironment (TME) [16][17].
2. Glucose Metabolism in Urothelial Carcinoma
Glucose metabolism produces energy in the form of ATP and precursor metabolites used for biosynthesis. Glucose metabolism starts with a process called glycolysis that consists of stepwise conversions of glucose that ultimately generates pyruvate. The rate of glycolysis is regulated by hexokinase (HK), glucose-6-phosphate dehydrogenase (G6PD), phosphofructokinase (PFK), and pyruvate kinase (PK) (Figure 1). Pyruvate participates in the tricarboxylic acid (TCA) cycle, also known as the citric acid cycle (CAC), or Krebs cycle, where it is ultimately oxidized into water and carbon dioxide. This oxygen-dependent process is called oxidative phosphorylation and ultimately produces 32–38 ATP molecules from one glucose molecule.
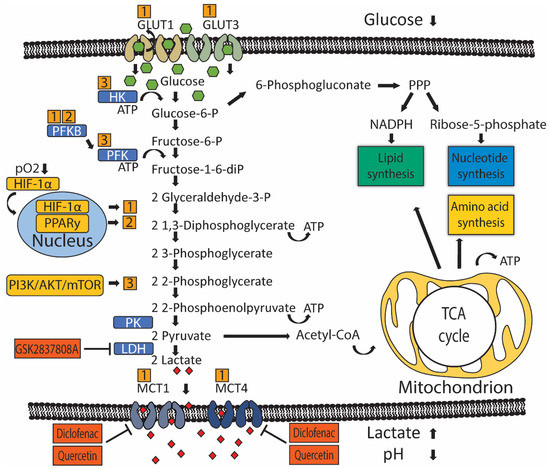
Figure 1. Metabolic reprogramming of glucose metabolism in UC. Oxygen-dependent hypoxia-inducible factor 1 alpha (HIF-1α) and oxygen-independent PI3K/AKT/mTOR and PPARy signaling (yellow boxes) drive metabolic reprogramming in UC by controlling the activity of rate-limiting enzymes (blue boxes) or transporters. Numbers (orange boxes) indicate which gene drives activation of particular rate-limiting enzymes or transporters. Glucose is imported into the cell by glucose transporters (GLUT) 1 and 3. Glycolysis starts with phosphorylation of glucose by hexokinase (HK) to glucose-6-phosphate, preventing glucose from diffusing outside the cell. Glucose-6-phosphate can be dehydrogenated by glucose-6-phosphate dehydrogenase (G6PD) to enter the pentose phosphate cycle (PPP) to produce pentoses like ribose-5-phosphate required for the synthesis of nucleotides and NADPH, which is necessary for reductive processes such as lipid biosynthesis. If glucose-6-phosphate is not oxidized by G6PD, glucose-6-phosphate is isomerized by phosphoglucose isomerase/phosphoglucoisomerase (PGI) to fructose-6-phosphate. Next, phosphofructokinase (PFK) catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-biphosphate, which irreversibly channels the glucose-derived metabolite into the glycolytic pathway towards phosphoenolpyruvate. At the end of the glycolytic pathway, pyruvate kinase (PK) catalyzes the dephosphorylation of phosphoenolpyruvate (PEP) to produce one pyruvate and one ATP molecule. Pyruvate is then metabolized into lactate-by-lactate dehydrogenase (LDH). Lactate is transported outside the cell by monocarboxylate transporters (MCT) 1 and 2, which leads to an increased concentration of extracellular lactate and an acidified tumor micro-environment. Pyruvate can also be metabolized into acetyl coenzyme A (acetyl-CoA), which participates in the tricarboxylic acid (TCA) cycle inside mitochondria to give rise to ATP and intermediary metabolites that are required for lipid and amino acid biogenesis.
In the absence of oxygen, pyruvate is transformed into lactate, producing 2 ATP molecules for every glucose molecule. In cancer cells, metabolic reprogramming (MR) refers to an upregulation of biosynthetic and bioenergetic pathways to produce the necessary materials and energy required for tumor growth. MR includes a shift in glucose metabolism from oxidation to glycolysis despite the presence of oxygen. This is commonly known as aerobic glycolysis [18] or the Warburg effect and favors the usage of glucose's carbon atoms for gaining biomass (i.e., metabolic building blocks) over energy (i.e., ATP) production. Because cancer cells also need more energy, yet this arrangement is energy inefficient, there is a compensatory increase in glucose consumption [19]. Glucose uptake is seen in UC by way of positron emission tomography/computed tomography (PET/CT), which uses radioactively labeled glucose-analog fluorodeoxyglucose (18F-FDG) to visualize primary tumors and metastases [3]. Human UC cell lines also show increased uptake of glucose compared to untransformed urothelial cells and produce increased levels of pyruvate and lactate [20]. Evaluation of the patient's UC tumor samples indicates glucose quantity was significantly lower compared to normal urothelium [21]. Furthermore, late TCA cycle intermediates were also increased in UC, suggesting flux into the TCA cycle to replenish intermediates extracted from the cycle for biosynthesis—a process called anaplerosis [21]. A significant increase of ribose, the end-product of the pentose phosphate pathway (PPP), was also observed in UC, suggesting upregulation of the PPP [21]. The PPP occurs in the cytosol and consists of an oxidative phase that produces NADPH, which is required for reductive processes such as fatty acid synthesis and scavenging of reactive oxygen species, and a non-oxidative phase that produces pentoses like ribose, which are important precursors for nucleotide synthesis. Therefore, the PPP helps metabolically active or proliferating cells to meet their anabolic demands and combat oxidative stress [22][23]. Thus, UC alters its metabolism and consumes glucose to produce energy via glycolysis, biomass through PPP and anaplerosis, and to counter oxidative stress through PPP.
3. Regulation of Glucose Transport and Metabolism in UC
Cellular glucose utilization is regulated by oxygen-dependent and oxygen-independent mechanisms that rely on several common glucose transporters and glycolytic enzymes (Figure 1). Oxygen-dependent mechanisms are mediated by transcription factor hypoxia-inducible factor 1-alpha (HIF-1α) [24][25]. Low oxygen tension stabilizes HIF-1α protein expression, which translocates to the nucleus and binds to target genes, thereby upregulating gene expression [26][27]. HIF-1α indirectly stimulates glycolysis through inhibition of mitochondrial biogenesis and oxygen consumption through induction of pyruvate dehydrogenase kinase 1 (PDK1), which subsequently inhibits pyruvate dehydrogenase from catalyzing oxidative decarboxylation of pyruvate [28][29]. The steroid receptor coactivator-3 (SRC-3) is a HIF-1α co-activator required for the expression of several HIF1-1α target genes in T24 UC cells under hypoxia [30]. Another HIF-1α co-activator is histone demethylase JMJD1A, whose H3K9 demethylase activity is required at promotor sites to induce expression of several key glycolytic enzymes [31]. Interestingly, JMJD1A was found upregulated in 46 UC patient samples, compared to 14 normal bladder samples [31]. In summary, UC has higher levels of HIF-1α co-activators, which leads to more glycolysis and reduced oxidative phosphorylation.
Oxygen-independent mechanisms of glucose utilization in UC are primarily mediated through activation of the PI3K/AKT/mTOR pathway [32][33]. The PI3K/AKT/mTOR pathway consists of activators: phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), mammalian target of rapamycin (mTOR), and PI3K-inhibitor: phosphatase and tensin homolog (PTEN). Mutations in genes of the PI3K/AKT/mTOR pathway are present in 42/131 (38%) patients with MIUC [34]. Besides activating mutations, other factors may also promote PI3K/AKT/MTOR signaling in UC. For example, microRNA 21 (Mir-21) activates PI3K/AKT/mTOR signaling through inhibition of PTEN expression in UC cell line T24, thereby stimulating glycolysis [32][35]. Furthermore, long non-coding RNA UCA1 is associated with mTOR-mediated glucose consumption and lactate production in 5637 human bladder carcinoma cells, although no direct interaction between mTOR and UCA1 was demonstrated [33].
Peroxisome proliferator-activated receptor gamma (PPARy) has been implicated as a driver of oxygen-independent activation of glycolysis in breast cancer and hepatocellular carcinoma murine models through transcriptional activation of key glycolytic enzymes [36][37]. In UC, the increased transcriptional activity of PPARy was associated with increased mRNA expression of glycolytic enzymes and decreased recurrence-free survival in a subset of 140 non-invasive (pTa) bladder tumors [38]. Interestingly, PPARy, like PI3K/AKT/mTOR signaling, is commonly associated with the luminal subtype of MIUC, which has a relatively good prognosis [39][40][41].
Increased glycolytic flux is associated with increased glucose uptake by glucose transporters. Glucose transporter 1 (GLUT1) is the primary glucose transporter overexpressed in cancer [42]. Expression of GLUT3 has also been demonstrated in T24 UC cells [43][44]. GLUT1 protein overexpression was associated with worse overall and disease-free survival in a pooled analysis of 4079 patients with various tumor types, not including UC [45]. GLUT1 expression evaluated by immunohistochemistry (IHC) in 105 BC samples was associated with an increased grade in both NMIUC and MIUC [46]. Furthermore, GLUT1 overexpression by IHC was an independent predictor of survival following radiotherapy (N = 64) or radical cystectomy (N = 279) for MIUC [47][48]. GLUT1 expression is generally induced by HIF-1α, indicating oxygen-dependent GLUT1 expression [49]. Likewise, GLUT1 and GLUT3 expression seem also to be controlled by microRNAs that function through altering PI3K/AKT/mTOR signaling in vitro [32]. Mir-218 was found to repress GLUT1 expression and, as a consequence, glucose uptake in T24 cells, while Mir-195-5p did the same for GLUT3 [43][44]. GLUT1 knockdown elevated intracellular reactive oxygen species (ROS) and increased cisplatin sensitivity in T24 cells [43].
Once glucose is imported into the cell, hexokinase (HK) (Figure 1) is the first rate-limiting enzyme controlling glycolytic flux. HK has four isoforms characterized by different functions and cellular locations. Isoform HK2 is linked to an anabolic function through PPP and has been implicated in UC MR [33][50][51]. T24 cells overexpress HK2 in response to PI3K/AKT/mTOR signaling [32]. Pharmacological inhibition of HK2 in UC cell line UM-UC-3 lowered glucose consumption and lactate production, accentuating a potential role in UC glucose metabolism [52].
UC cells also have upregulated phosphofructokinase (PFK) (Figure 1), which drives increased glycolytic flux. Somatic genetic aberrations that upregulate or amplify PFK family genes are present in ~40% of MIUC patients [50]. In vitro studies with UC cell lines, RT4, and TCCSUP suggested that PFK is primarily important during early phases of cancer progression, as PFK expression was higher in RT4 (representing early-stage, well-differentiated NMIUC) compared to TCCSUP (representing more progressed, anaplastic MIUC) [51]. Moreover, a lower PFK activity was associated with increased pyruvate consumption, implying that more progressed tumors start to directly metabolize pyruvate instead of glucose [51]. PFK is indirectly activated by one of four 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB) enzymes. PFKFB3 is expressed in T24, and knockdown led to decreased lactate production [53]. Another PFKFB family member, PFKFB4, was found expressed in 135 UC radical cystectomy samples [54]. High PFKFB4 expression assessed by IHC was associated with increased tumor stage and grade, and subsequent in vitro experiments demonstrated that PFKFB4 expression was induced during hypoxia in an HIF-1α-dependent manner [54].
The last step of glycolysis converts phosphoenolpyruvate (PEP) and ADP to pyruvate and ATP, and this step is catalyzed by pyruvate kinase (PK) isozymes M1/M2 (PKM1/M2) (Figure 1). UC cell lines have been shown to reexpress PKM2 [55].
In cancer cells, pyruvate is metabolized to lactate-by-lactate dehydrogenase (LDH), which reduces NADH to NAD+ in the same process. Lactate production replenishes cytosolic NAD+, allowing a continuous glycolytic flux [56][57]. Lactate produced by LDH is exported across the cell membrane by monocarboxylate transporters (MCT) in order to maintain an alkaline intracellular pH, favoring metabolism [58][59]. Tumor cells depend on MCT4 and, to a lesser extent, on MCT1 for lactate export, and MCT4 is expressed in a HIF-1α dependent manner [59][60]. MCT4 was overexpressed in approximately 50% of 360 UC patients, as assessed by IHC [61]. Moreover, MCT4 protein overexpression was an independent prognostic factor, predicting poor recurrence-free survival in NMIUC and MIUC patients treated with transurethral resection or radical cystectomy [61]. Likewise, MCT4 mRNA and protein expression predicted poor overall survival in MIUC patients treated with radical cystectomy [62]. Short interference RNA (siRNA) mediated silencing of MCT4 in UC cell lines, reduced proliferation rates, and increased ROS in a glucose-dependent manner [62]. Moreover, stable shRNA knockdown of MCT4 impaired tumor growth in an orthotopic UC xenograft model [62].
In conclusion, evidence shows that UC uses HIF-1α to increase glycolytic flux and to neutralize ROS via the upregulated activity of glucose importers (GLUT1, GLUT3), glycolytic enzymes (PFK), and lactate transporters (MCT4). Meanwhile, PI3K/AKT/mTOR signaling contributes to upregulating glycolytic enzymes (HK). Inhibiting glycolysis and lactate production may target UC either directly by impairing metabolic activity. However, most mechanistic evidence was gathered in small studies investigating parts of UC metabolism in a few human UC cell lines. More comprehensive preclinical investigation of UC metabolism in different stages of the disease is needed to increase the validity of these findings before translation into clinical trials.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN esti-mates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424, doi:10.3322/caac.21492.
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657, doi:10.1016/j.eururo.2019.08.016.
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020, doi:10.1016/j.eururo.2020.03.055.
- Von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077, doi:10.1200/JCO.2000.18.17.3068.
- Dash, A.; Galsky, M.D.; Vickers, A.J.; Serio, A.M.; Koppie, T.M.; Dalbagni, G.; Bochner, B.H. Impact of renal impairment on eligibility for adjuvant cisplatin-based chemotherapy in patients with urothelial carcinoma of the bladder. Cancer 2006, 107, 506–513.
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.; Sternberg, C.N.; Bajorin, D.F.; et al. Treatment of Patients With Metastatic Urothelial Cancer “Unfit” for Cisplatin-Based Chemo-therapy. J. Clin. Oncol. 2011, 29, 2432–2438, doi:10.1200/jco.2011.34.8433.
- Galsky, M.D.; Chen, G.J.; Oh, W.K.; Bellmunt, J.; Roth, B.J.; Petrioli, R.; Dogliotti, L.; Dreicer, R.; Sonpavde, G. Compara-tive effectiveness of cisplatin-based and carboplatin-based chemotherapy for treatment of advanced urothelial carcinoma. Ann. Oncol. 2012, 23, 406–410.
- Von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608, doi:10.1200/JCO.2005.07.757.
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11, doi:10.1038/s12276-018-0191-1.
- Powles, T.; Duran, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castella-no, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or meta-static urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757.
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.O.; Bra-carda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322, doi:10.1016/s1470-2045(17)30065-7.
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026.
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492.
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16, doi:10.1038/bjc.2017.434.
- Pitt, M.J.; Vétizou, M.; Daillère, R.; Roberti, María, P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, Ivo, G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and Extrinsic Factors. Immunity 2016, 44, 1255–1269, doi:10.1016/j.immuni.2016.06.001.
- Afonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder can-cer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106, doi:10.1038/s41585-019-0263-6.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029, doi:10.1126/science.1160809.
- Whyard, T.; Waltzer, W.C.; Waltzer, D.; Romanov, V. Metabolic alterations in bladder cancer: Applications for cancer imaging. Exp. Cell Res. 2016, 341, 77–83.
- Sahu, D.; Lotan, Y.; Wittmann, B.; Neri, B.; Hansel, D.E. Metabolomics analysis reveals distinct profiles of nonmus-cle-invasive and muscle-invasive bladder cancer. Cancer Med. 2017, 6, 2106–2120.
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354.
- Hsu, M.M.; Balar, A.V. PD-1/PD-L1 Combinations in Advanced Urothelial Cancer: Rationale and Current Clinical Trials. Clin. Genitourin Cancer 2019, 17, e618–e626, doi:10.1016/j.clgc.2019.03.009.
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Na-ture 2006, 441, 437–443.
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet Dev. 2010, 20, 51–56, doi:10.1016/j.gde.2009.10.009.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197, doi:10.1016/j.cmet.2006.01.012.
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185, doi:10.1016/j.cmet.2006.02.002.
- Zhao, W.; Chang, C.; Cui, Y.; Zhao, X.; Yang, J.; Shen, L.; Zhou, J.; Hou, Z.; Zhang, Z.; Ye, C.; et al. Steroid receptor co-activator-3 regulates glucose metabolism in bladder cancer cells through coactivation of hypoxia inducible factor 1α. J. Biol. Chem. 2014, 289, 11219–11229.
- Wan, W.; Peng, K.; Li, M.; Qin, L.; Tong, Z.; Yan, J.; Shen, B.; Yu, C. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene 2017, 36, 3868–3877.
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 dimin-ishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour. Biol. 2015, 36, 383–391.
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955.
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322, doi:10.1038/nature12965.
- Tao, J.; Lu, Q.; Wu, D.; Li, P.; Xu, B.; Qing, W.; Wang, M.; Zhang, Z.; Zhang, W. microRNA-21 modulates cell prolifera-tion and sensitivity to doxorubicin in bladder cancer cells. Oncol. Rep. 2011, 25, 1721–1729, doi:10.3892/or.2011.1245.
- Shu, Y.; Lu, Y.; Pang, X.; Zheng, W.; Huang, Y.; Li, J.; Ji, J.; Zhang, C.; Shen, P. Phosphorylation of PPARγ at Ser84 pro-motes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget 2016, 7, 76984–76994, doi:10.18632/oncotarget.12764.
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARγ in breast cancer pathophysiology. J. Drug Target 2013, 21, 161–174.
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715.e707, doi:10.1016/j.ccell.2017.08.005.
- Rochel, N.; Krucker, C.; Coutos-Thévenot, L.; Osz, J.; Zhang, R.; Guyon, E.; Zita, W.; Vanthong, S.; Hernandez, O.A.; Bourguet, M.;; et al. Recurrent activating mutations of PPARγ associated with luminal bladder tumors. Nat. Commun. 2019, 10, 253–253, doi:10.1038/s41467-018-08157-y.
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433, doi:10.1016/j.eururo.2019.09.006.
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e525, doi:10.1016/j.cell.2017.09.007.
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26, doi:10.4067/s0716-97602002000100004.
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell Physiol. Biochem. 2017, 41, 921–932.
- Fei, X.; Qi, M.; Wu, B.; Song, Y.; Wang, Y.; Li, T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012, 586, 392–397, doi:10.1016/j.febslet.2012.01.006.
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367.
- Reis, H.; Tschirdewahn, S.; Szarvas, T.; Rübben, H.; Schmid, K.W.; Grabellus, F. Expression of GLUT1 is associated with increasing grade of malignancy in non-invasive and invasive urothelial carcinomas of the bladder. Oncol. Lett. 2011, 2, 1149–1153.
- Hoskin, P.J.; Sibtain, A.; Daley, F.M.; Wilson, G.D. GLUT1 and CAIX as intrinsic markers of hypoxia in bladder cancer: Relationship with vascularity and proliferation as predictors of outcome of ARCON. Br. J. Cancer 2003, 89, 1290–1297, doi:10.1038/sj.bjc.6601260.
- Boström, P.J.; Thoms, J.; Sykes, J.; Ahmed, O.; Evans, A.; van Rhijn, B.W.; Mirtti, T.; Stakhovskyi, O.; Laato, M.; Margel, D.; et al. Hypoxia Marker GLUT-1 (Glucose Transporter 1) is an Independent Prognostic Factor for Survival in Bladder Cancer Patients Treated with Radical Cystectomy. Bladder Cancer 2016, 2, 101–109.
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192, doi:10.1038/nrc2344.
- Sun, C.M.; Xiong, D.B.; Yan, Y.; Geng, J.; Liu, M.; Yao, X.D. Genetic alteration in phosphofructokinase family promotes growth of muscle-invasive bladder cancer. Int. J. Biol. Markers 2016, 31, e286–e293.
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The progres-sion from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and py-ruvate metabolism. Exp. Cell Res. 2015, 335, 91–98.
- Lin, H.; Zeng, J.; Xie, R.; Schulz, M.J.; Tedesco, R.; Qu, J.; Erhard, K.F.; Mack, J.F.; Raha, K.; Rendina, A.R.; et al. Discov-ery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217–222.
- Hu, K.Y.; Wang de, G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951.
- Zhang, H.; Lu, C.; Fang, M.; Yan, W.; Chen, M.; Ji, Y.; He, S.; Liu, T.; Chen, T.; Xiao, J. HIF-1α activates hypoxia-induced PFKFB4 expression in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2016, 476, 146–152.
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Blad-der Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983.
- Massari, F.; Ciccarese, C.; Santoni, M.; Iacovelli, R.; Mazzucchelli, R.; Piva, F.; Scarpelli, M.; Berardi, R.; Tortora, G.; Lopez-Beltran, A.; et al. Metabolic phenotype of bladder cancer. Cancer Treat Rev. 2016, 45, 46–57.
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2004, 447, 619–628, doi:10.1007/s00424-003-1067-2.
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 2011, 226, 299–308, doi:10.1002/jcp.22400.
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349.
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037, doi:10.1074/jbc.M511397200.
- Choi, J.-W.; Kim, Y.; Lee, J.-H.; Kim, Y.-S. Prognostic Significance of Lactate/Proton Symporters MCT1, MCT4, and Their Chaperone CD147 Expressions in Urothelial Carcinoma of the Bladder. Urology 2014, 84, 245.e215–245.e249, doi:10.1016/j.urology.2014.03.031.
- Todenhöfer, T.; Seiler, R.; Stewart, C.; Moskalev, I.; Gao, J.; Ladhar, S.; Kamjabi, A.; Al Nakouzi, N.; Hayashi, T.; Choi, S.; et al. Selective Inhibition of the Lactate Transporter MCT4 Reduces Growth of Invasive Bladder Cancer. Mol. Cancer Ther. 2018, 17, 2746–2755.


