 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ramin Zibaseresht | + 712 word(s) | 712 | 2021-02-11 12:08:27 | | | |
| 2 | Catherine Yang | -108 word(s) | 604 | 2021-02-20 04:27:28 | | |
Video Upload Options
Synthesis strategy of a terpyridine bridging ligand was explained.
4'-(p-(N,N′-bis(2-pyridylmethyl)amine)-N"-methylphenyl)-2,2':6',2"-terpyridine (bpat)
1. Introduction
We have begun a research programme aimed at the synthesis of heterodinuclear complexes. In these studies, we are focusing on the preparation and use of ditopic ligands where the two metal ion binding sites are differentiated by the number of donor atoms in each site, the configuration of the binding site, or the types of donor atom that are present in the sites. This binding site differentiation offers the prospect of using the different coordination properties of the binding sites to control the regiochemistry of complexation, ensuring that the correct metal ion is incorporated at the correct binding site in the ligand.
2. Application
In order to prepare heterodinuclear metal complexes, or a variety of inorganic polymers, the ligand acts as linker between the metal atoms. Those who are particularly interested in minimising the number of stereoisomers that might be produced on synthesis of the heterodinuclear systems, this type of linker can be one of their choices.
3. Ligand Synthesis
Ditopic ligands 4'-(p-(1,5-bis(phthalimido)-3-azapentan-3-yl)methylphenyl)-2,2':6',2"-terpyridine (bpat), based on tridentate planar terpyridyl systems has been synthesised in relatively high yield. The ligand was prepared using the method by nucleophilic substitution reaction of 4’-(p-bromomethylphenyl)-2,2’:6’,2’’-terpyridine with the appropriate secondary or tertiary amines in single- or multi-step reactions.
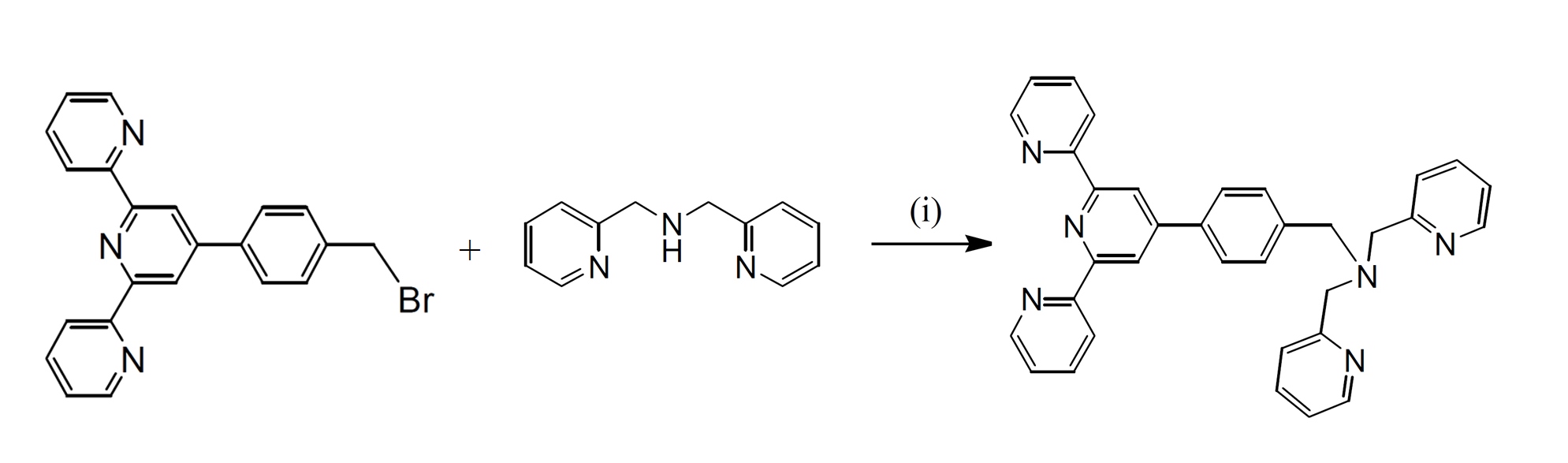
Scheme 1. Syntheses of ligand bpat. (i) dipicolylamine (N,N’-bis(2-pyridylmethyl)amine), K2CO3, dry CH3CN, 3 days, 60o C
Ligand bpat was synthesised using direct functionalisation of dipicolylamine (N,N’-bis(2-pyridylmethyl)amine) with the bromo compound. The reactions were carried out in dry CH3CN and in the presence of K2CO3 to afford the desired products in excellent yields (95-98%). The reaction mixtures were monitored by 1H NMR spectroscopy and thin layer chromatography (TLC) in order to determine the optimum conditions. It was noted that prolonged reactions (up to 3 days) and also using 1.2 equivalents of the amines resulted in analytically pure products, with the best yields. The product was characterised using 1H and 13C NMR spectroscopy, COSY, NOESY, HSQC, and electrospray ionisation mass spectrometry.
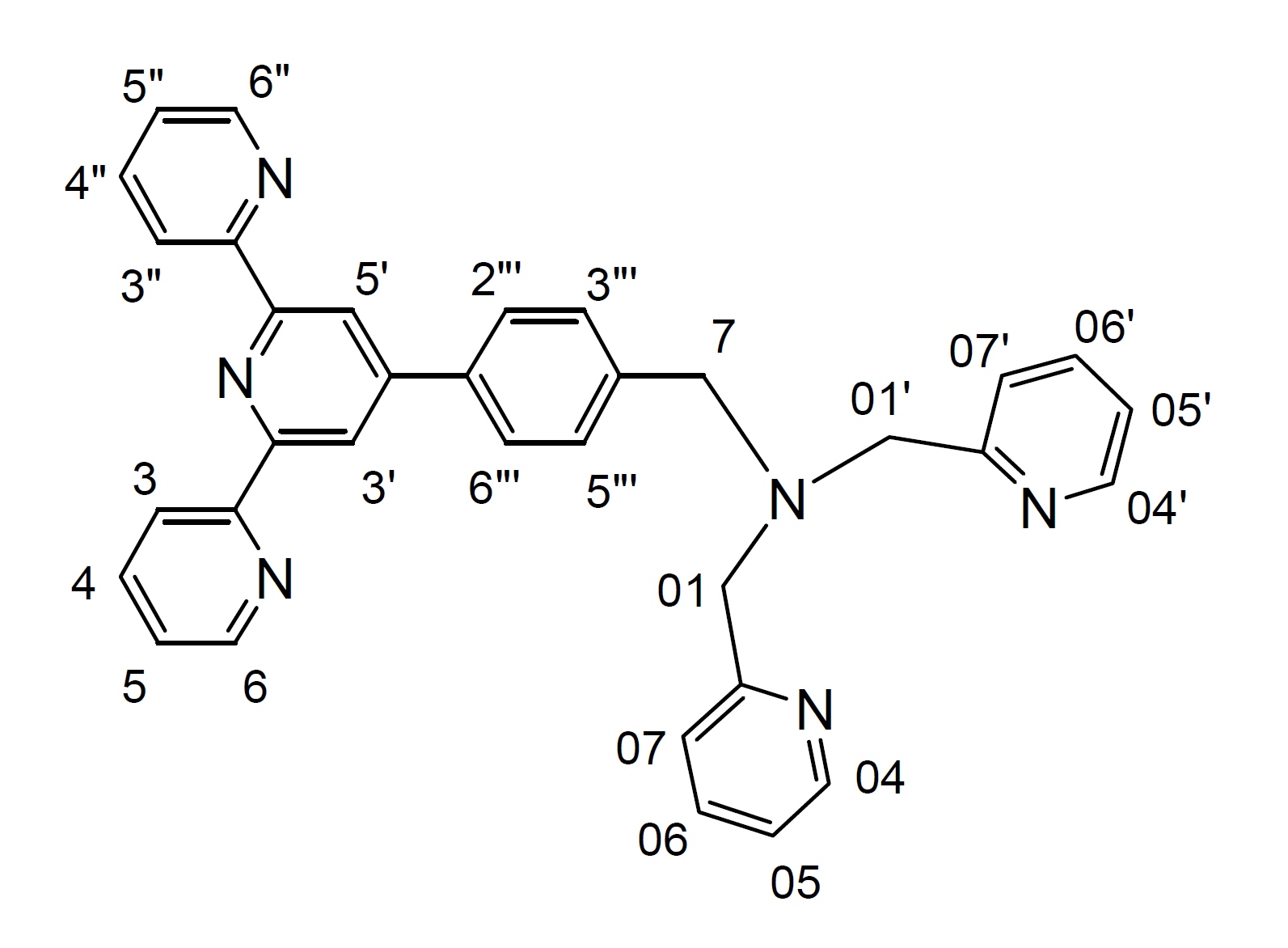
A mixture of (N,N'-bis(2-pyridylmethyl)amine), (0.24 g, 1.2 mmol) and 4'-(p-bromomethylphenyl)-2,2':6',2"-terpyridine (btp) (0.4 g, 1.0 mmol), and anhydrous K2CO3 (1.4 g, 10 mol) in dry CH3CN (50 mL) was stirred at 55-60° C under argon for 3 days. The progress of the reaction was monitored by TLC. The reaction mixture was then allowed to cool at room temperature; filtered and the solvent was removed from the filtrate under reduced pressure to give a pale yellow oil. Purification by column chromatography (Al2O3, 1-2% MeOH in CH2Cl2) afforded the desired product (0.52 g, 96%) as a pale yellow oil. 1H NMR (500 MHz; solvent CDCl3): δ 8.729 (2H, s, H3', H5'), 8.725 (2H, d, H6, H6"), 8.67 (2H, d, H3, H3"), 8.54 (2H, d, H04, H04'), 7.87 (2H, m, H4, H4"), 7.86 (2H, d, H2"', H6"'), 7.69 (2H, m, H06, H06'), 7.61 (2H, d, H07, H07'), 7.56 (2H, d, H3"', H5"'), 7.35 (2H, m, H5, H5"), 7.17 (2H, m, H05, H05'), 3.86 (4H, s, H01, H01'), 3.77 (2H, s, H7). 13C NMR (75 MHz; solvent CDCl3): δ 159.61, 156.24, 155.87, 150.03, 149.09 (2C, C6, C6"), 148.98 (2C, C04, C04'), 140.18, 137.23, 136.83 (2C, C4, C4" or C2"', C6"'), 136.46 (2C, C06, C06'), 129.35 (2C, C3"', C5"'), 127.26 (2C, C4, C4" or C2"', C6"'), 123.78 (2C, C5, C5"), 122.85 (2C, C07, C07'), 122.00 (2C, C05, C05'), 121.31 (2C, C3, C3"), 118.74 (2C, C3', C5'), 60.06 (2C, C01, C01'), 58.20 (C7). ESI-MS: m/z 261.1370 ([M+2H]2+, 100%), 521.2880 ([M+H]+, 14%).

