 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlos Monteiro | + 3214 word(s) | 3214 | 2021-02-09 03:54:05 | | | |
| 2 | Catherine Yang | -4 word(s) | 3210 | 2021-02-20 10:10:21 | | |
Video Upload Options
Since the serendipitous discovery of phthalocyanines (Pcs), at the beginning of the 20th century, growing attention has been devoted to the ubiquitous catalytic properties and applications of Pcs and Pcs coordinated with metal ions, Metallophthalocyanines (MPcs). In fact, these old synthetic dyes have been very attractive in the view of academic research, but mainly for catalytic industrial purposes as these dyes are easily accessible on a large scale, are robust under harsh reaction conditions, show high activity, and can be recovered and recycled. The success and diversity of MPcs as catalysts over different substrates are mostly attributed to their high structural flexibility in terms of metals and electronic properties of the phthalocyanine ligands.
1. General Introduction
After the casual discovery of phthalocyanines (Pcs) at the beginning of the 20th century [1], these macrocycles have been the subject of deep studies, mainly focused on their valuable dye properties [2]. Along the first decades of 20th century, the importance of the coordination of Pcs with metal ions in their synthesis, and in the tuning of their chemical and dyeing properties was explored and emphasized [3][4]. In more recent years, the role of Pcs and their metal complexes (MPcs) as industrial dyes and pigments, was accompanied by an intensive search aiming to develop new fields of application [5][6]. In particular, a growing attention is focused on the redox catalytic properties of MPcs [7]. Although non-natural, Pcs show a noticeable resemblance to the structurally related macrocycle porphyrin, and also to other non‑natural macrocycles such as porphyrazines and tetrabenzoporphyrins (Figure 1).
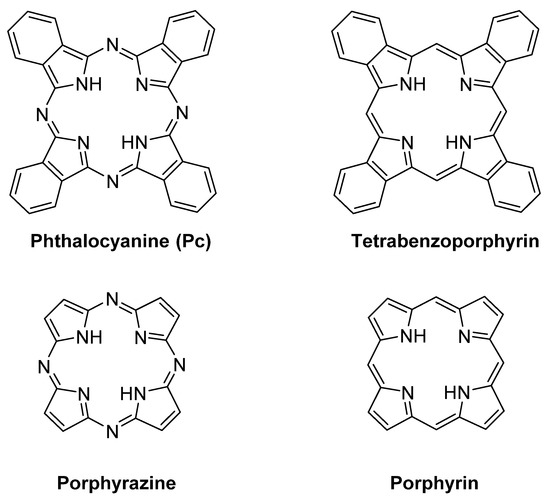
Figure 1. Chemical structures of tetrapyrrolic macrocycles: phthalocyanine, tetrabenzoporphyrin, porphyrazine, and porphyrin core.
Like porphyrinoids, Pcs are aromatic macrocycles obeying the Hückel’s rule (with 18 delocalized π electrons). However, the UV–Vis spectral patterns clearly reveal sharp differences that are reflected in the general chemical properties. Rather than from the benzocondensation, such differences mainly arise from the tetra-aza substitution of the four methinic bridges joining the pyrrolic-type rings in porphyrins. The four additional nitrogen atoms exert an electron-withdrawing effect, but also bear filled non-bonding orbitals, which are responsible for n→π transitions, increasing the width of the main absorption bands [8]. In fact, the Q bands of Pcs arise from a1u to eg transitions, whereas in porphyrins the corresponding bands are due to a2u to eg transitions [9].
Pcs show a high appetency to form the corresponding dianions upon deprotonation under suitable conditions, affording in the presence of the adequate metal ions the corresponding metal complexes MPcs (Figure 2).

Figure 2. Chemical structures of metallophthalocyanine complexes (MPcs) with different metals.
2. Metallophthalocyanines as Redox Catalysts: An Overview
As noted above, the first noteworthy property of MPcs was their chemical “inertness”, which made and make them very attractive as stable and durable industrial dyes. However, their rich redox chemistry has only been explored in the last few decades [10].
In fact, MPcs are protected by their structure against the main threats jeopardizing redox‑active metalloporphyrins, and that is the outstanding limitation towards an extensive use of the latter as industrial catalysts. First, the methinic bridges joining the pyrrole-type rings in porphyrins are the preferential targets of oxidative attack, leading initially to oxophlorins and later to macrocycle breaking and therefore to catalyst destruction [11]. Such methinic bridges are replaced in MPcs by aza bridges, which obviously are generally inert towards oxidative attacks. Second, the four condensed benzene rings protect the pyrrolic β-positions against any unwanted substitution reaction and prevent any possibility of pyrrole ring cleavage. However, MPcs are not totally indestructible, and some reports exist, describing their degradation under relatively mild reaction conditions [12][13][14]. Nevertheless, in certain cases, the degradation products still show catalytic activity [15].
The Pc anions (Pc2−) forming the various MPcs can be oxidized by one- or two-electron processes, leading, respectively, to radical anions Pc•− or to a neutral species Pc. Conversely, reduction by one to four electrons affords, respectively, Pc3−, Pc4−, Pc5−, and Pc6−. Obviously, Pc3− and Pc5− have a radical character. Most of these oxidized or reduced species could be formed by means of electrochemical methods [10] and some are not found along the “normal” MPc chemistry. The central metal ion may be not involved in redox processes, as it can be anticipated for the majority of the main groups elements and for some transition metals such as Ni(II). However, in the case of most transition metals—they can show a rich redox chemistry while bound to the Pc ligand, which in turn is subject to its own redox changes.
A judicious choice of the peripheral substituents [16] in MPcs easily affords very soluble compounds, either in organic solvents or in water, suitable for electrochemical studies, or for several applications as redox catalysts. Moreover, the axial coordination to the central metal ions in the Pc inner core is responsible by further changes in their electrochemistry as well as in their redox properties, thus opening the way to a huge number of different redox-active species, that in principle could find application as redox catalysts. In this regard, MPcs show a large versatility, being able to work as photocatalysts, as electrocatalysts, and as “classical” catalysts, by activating oxidizing species such as O2, H2O2, t-BuOOH, KHSO5, and so on. MPc-mediated electrocatalysis and photocatalysis are out of the scope of the present minireview, whereas oxidative catalysis with oxygen will be reviewed and discussed.
Stillman and Nyokong [17] have reviewed some general (and in particular spectroscopic) properties of nearly all phthalocyanine complexes with metals and also nonmetals, with additional focus on their redox properties, when the central metal is directly involved. Generally speaking, some MPcs, such as MnPcs, FePcs, or CoPcs, could directly interact, in their lower oxidation states, with dioxygen, forming adducts, more or less resembling the corresponding ferrohemoprotein-dioxygen adducts, such as oxyhemoglobins, oxymyoglobins, and also the ferroheme-containing cytochromes P-450 intermediate adducts with dioxygen. Some of these such adducts, which could be to a certain extent reversible, have been individualized; others should be transient species, more or less rapidly evolving to more stable products, where the oxidation state of the central metal ion has become higher. Comparative studies dealing with the ease of formation of such dioxygen adducts, and their tendency to undergo electroreduction, are those of Wang et al. [18] and of Shi and Zhang [19] that used Density Functional Theory to predict the electrocatalytic ability of some FePc and CoPc complexes in solution. Although specifically aimed to electrocatalysis, these studies are a suitable theoretical base to infer information about the catalytic redox properties of those phthalocyanines metal complexes and to extend the conclusions to similar ones.
As noted above, such adducts could arise from direct reaction of the corresponding MPcs, containing a proper divalent metal cation, with dioxygen. However, their similarity with ferroheme-dioxygen adducts is substantially low, as they are in fact superoxide complexes of the MPcs, where the oxidation number of the metal ions has increased to +3. Therefore, depending on the specific metal, on the peripheral substituents on the Pc macrocycle, and on the particular experimental conditions, they can act as one-electron oxidizers (where the oxidation number of the metal ion reverts to +2, whereas the superoxide more or less rapidly is degraded to H2O2). Alternatively, “direct” oxygenation of certain substrates could take place (vide infra).
MnPc and its substituted derivatives [20] can contain either Mn(II) and Mn(III); the very complex redox equilibria between the two oxidation states are highly sensitive to the presence of even traces of water. Additionally, reactions of Mn(II) derivatives with dioxygen (O2) are very sensitive to the experimental conditions. In absolutely dry solvents such as pyridine, Mn(II)Pc readily forms a monomeric adduct O2-MnPc which has the electronic arrangement O2−-Mn(III)Pc•+ corresponding to a superoxide adduct of Mn(III)Pc, which is the formal analogue of peroxidase Compound III (Figure 3) [21]. By contrast, when water traces are present, the reaction between dioxygen and Mn(II)Pc leads to a µ-oxo dimer PcMn(III)—O—Mn(III)Pc (Figure 3). With time, the µ-oxo dimer could also arise from the degradation of the Mn(III)-containing superoxide complex. Therefore, the superoxide adduct could well be the first, transitory reaction product, turning to further products under the influence of water traces. MnPcs, where eight electron-withdrawing substituents are present at the benzene rings, can exist as both stable Mn(II) and Mn(III) oxidation states, with no appreciable attitude to directly react with dioxygen. This feature opens the way to further studies, where the Mn(II)/Mn(III) complex couples could well exert interesting redox activities.
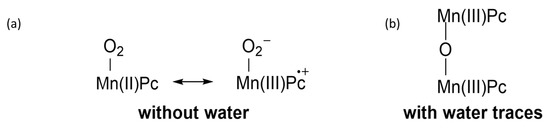
Figure 3. Electronic arrangements of Mn(II)Pc after reaction with dioxygen: (a) in the absence of water and (b) in the presence of water traces.
The experimental conditions have a great influence on the equilibria between Mn(II) and Mn(III) Pcs, as well as on their oxygenated derivatives; under certain conditions, the oxygenation to afford Mn(III)PcO2 is reversible [22]. The peculiar catalytic properties of such superoxide adducts have been reported many years ago, as it catalyzes the di-oxygenation (with concomitant ring cleavage) of some indole derivatives [23]. Interestingly, the reaction is rather specific: 3-methyl-, 2,3-dimethyl-indole, and tryptophan are substrates, whereas plain indole, 1-methyl-, 2-methyl-, 1,2-dimethyl-, and 1,3-dimethyl-indole are not. In conclusion, a substituent must be present at the 3-position, whereas the 1-position must be unsubstituted, and the 2-position has not influence towards the susceptibility of the compound to the catalytic di-oxygenation reaction. When it happens, the di-oxygenation invariably affects the 2,3-bond, and in the case of the simplest substrate, 3-methyl-indole, the reaction product is 2-formamido-acetophenone. Although no detailed mechanism was proposed for the reaction, there is evidence that at the end of each catalytic cycle Mn(II)Pc must be obtained, which in turn reacts again with dioxygen to produce the reactive superoxide adduct.
The behaviour of the MnPcs to cycle between the two oxidation states of the central metal ion opened the way to a number of studies about the redox catalytic properties of the manganese complexes of variously substituted—and very often water-soluble—phthalocyanines. Moxon and colleagues [24] studied in detail the dioxygen adduct of symmetrical tetrasulfonated-Mn(II)Pc, and some of its changes upon pH variations. First of all, the solid adduct contains manganese(III) and superoxide. This arrangement changes upon dissolving the adduct in water, where it changes to a Mn(II) complex, binding dioxygen in a manner formally resembling oxymyoglobin and oxyhemoglobin. When pH is raised to >11, an intramolecular redox reaction takes place, and the Mn(III) adduct of superoxide is formed. At pH values > 14, the complex is reversibly destroyed with dioxygen evolution, but the process can be reversed by lowering the pH, for example to ∼3, where the Mn(II) is formed again, for which the authors have also claimed an H2O2-forming oxidase activity. These results form the background for further studies on the suitability of MnPcs as redox catalysts which be discussed in detail.
Adducts arising from Fe(II)Pcs and O2 must exist as reactive intermediates, very quickly evolving to stable products, namely the μ-oxo dimer of the type PcFe(III)–O–Fe(III)Pc. In relation to such a reactivity of the primary adduct, different FePcs under different experimental conditions could participate as catalysts in a wide variety of aerobic oxidation or oxygenation reactions. Many years ago, Ercolani and colleagues [25], working on unsubstituted Fe(II)Pc, found that the putative O2-Fe(II)Pc complex, under suitable experimental conditions (toluene solution, high oxygen pressure), was able to catalyze the oxygenation of triphenylphosphine to the corresponding P-oxide. During the reaction, the insoluble Fe(III)-containing μ-oxo dimer slowly precipitated, with concomitant exhaustion of the catalysis. In the presence of two equivalents of pyridine, which promoted the formation of the soluble and catalytically very active Fe(II)(Py)2Pc complex, the efficiency increased, and for this reaction the authors proposed the formation of the transient ferryl species O=Fe(IV)(Py)Pc. However, also this complex slightly decomposed with formation of the μ-oxo dimer, but the excess of pyridine prevented the formation of the latter and consequent catalyst inactivation. Interestingly, under the same experimental conditions, both Mn(II)Pc and Co(II)Pc were found to be quite inactive, despite their ability to form dioxygen adducts.
Additionally, rather complicated is the chemistry of the CoPcs with O2, which in some cases requires the presence of H2O2 to afford the adduct formation [26]; the formation of a transient O2−-Co(III)Pc adduct has been suggested for other “autoxidation” reactions [27]; the behavior of non-supported Co(II)Pcs to readily form Co(III) µ‑peroxo dimers is a well-established fact, which can be prevented by proper axial coordination [28]. As a consequence of a mono-axial coordination, the O2-CoPc adducts become more efficient as oxidants. Of particular interest is the catalytic activity of Co(II)Pcs in aerobic oxidation of thiols to the corresponding disulfides, whose mechanism has been fully elucidated by Pan and coworkers (see Figure 16, as an example) [29]. The Co(II)Pc catalyst forms a ternary, bis-axial (octahedral) complex with the thiolate ion and dioxygen (the latter in the superoxide form, whereas the cobalt ion is in its +3 oxidation state). Then, a one-electron transfer to the cobalt(III) ion takes place, affording a thiyl radical RS• whereas the metal is reduced back to Co(II). This causes the release of superoxide anion, which in turns reacts very rapidly with another thiolate ion, leading finally to disulfide and H2O2 as the reaction products.
As noted above, another crucial feature of MPcs containing redox-active metal ions such as Fe, Co, Mn, Ru, and others, is their ability, in particular when in their +3 oxidation state, to react with peroxo compounds or other oxygen donors such as iodosobenzene and iodibenzene derivatives [30]. More frequently, H2O2 and its alky or acyl derivatives have been explored for their ability to form peroxo complexes or hypervalent derivatives of MPcs (Figure 4). In principle, some compounds should be formed upon reaction of a suitable M(III)Pc with a generic hydroperoxide R–O–OH: (1) a Compound zero analogue, RO–O–M(III)Pc; (2) a Compound I analogue O=M(V)Pc ↔ µ O=M(IV)Pc•+↔ µ O•–M(IV)Pc; (3) a Compound II analogue O=M(IV)Pc ↔ µ O•–M(III)Pc. As noted above, the electron-withdrawing effect of the phthalocyanine ring is, generally speaking, stronger than that observed for its porphyrin counterpart, and as a consequence the above reported compounds are more reactive but also more unstable [10]. This explains why until now only a few examples have been isolated and characterized, such as the inert O=V(IV)Pc [31][32] and O=Nb(IV)Pc [33], contrarily to that observed in the case of the corresponding metalloporphyrin compounds [34][35][36][37][38][39]. Apart from the formal resemblance between the porphyrin and Pc series, some substantial differences can be observed between the two families of redox-active metal complexes [7]. In fact, several different mechanisms can operate, as a function of the particular MPc catalyst, of the chosen oxidant, and of the particular substrate to be oxidized. The system Fe(III)Pc-BuOOH could operate with a one-electron mechanism—starting from the homolytic scission of the peroxo bridge O–O—when oxidizing phenols to quinones, although it oxidizes to quinones some other substrates such as anthracene and xanthene with a two-electron mechanism (as a consequence of a heterolytic scission releasing OH– and affording a very reactive hypervalent intermediate), as shown by the absence of oxidative coupling products, that are typical for one-electron (radical) mechanisms. On the other hand, the significant incorporation of 18O from 18O2 along the oxidation of alkynes is a convincing proof of the radical mechanism. The reasons for such varying behavior of this and other MPc catalysts is still unknown. The propensity of Fe(III)Pc complexes to remain in the +3 oxidation state of the Compound 0 analogues has been assessed and discussed many years ago [40] and confirmed more recently [41]. In fact, the anionic form of such complexes,–O–O–Fe(III)Pc, is an effective nucleophile acting as an epoxidizing agent towards electron-deficient double bonds, incorporated into an aromatic ring, for example in polychlorophenols [42].
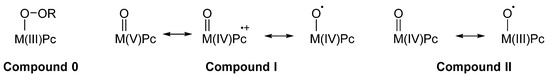
Figure 4. Complexes or hypervalent derivatives of MPcs formed upon reaction of a suitable M(III)Pc with a generic hydroperoxide R–O–OH.
An outstanding feature of some MPcs is their high tendency to form μ-bridged dimers, where the two metal ions are linked together by –O– (oxo), –N= (nitrido) or also =C= (carbido) bridges [43]. Among these, diiron-oxo-bridged phthalocyanines have been deeply studied, mainly due to the easily preparation [7], as well as other binuclear complexes, containing different metal ions such as Ru, Cr, Mn, in different combinations (homo- or hetero-metallic) eventually hosted in the same Pc or in different Pcs (homo- or hetero-leptic complexes) [44]. In fact, when such a complex is treated with a peroxo compound, a Compound 0 analogue, involving only one Fe(III), is formed. This evolves preferentially to a Compound I analogue, as the formal positive charge arising on the involved Fe ion is efficiently delocalized, through the μ-oxo bridge, on the other Fe ion. In this way, a O=Fe(IV)Pc–O–Fe(IV)Pc is formed, lacking both the radical cation character and the positive charge, inherent to the Compound I analogue arising from the monometallic counterpart [41]. As a consequence, the hypervalent oxo compound deriving from the binuclear complex represents the optimal balance between stability against auto-destruction and effectiveness of oxidation catalysis. In other words, the two-electron oxidation mechanism arising from the heterolytic cleavage of the –O–O– peroxo bridge in the binuclear complex affords selective oxidation pathways for many to-be-oxidized substrates, in contrast with the mononuclear counterpart, where the homolytic cleavage leads to radical mechanism with concomitant oxidant wasting and catalyst consumption. Unluckily, along the catalysis the binuclear complex could gradually decompose into the mononuclear species, therefore lowering the overall catalytic efficiency [41][45]. The immobilization of the binuclear complexes on to suitable insoluble supports could obviate to this drawback, when a reaction medium is chosen, unable to dissolve the formed mononuclear species.
The things go quite differently in the case of binuclear complexes where the two metal ions are linked by a nitrido bridge. In fact, these compounds, whose importance in catalysis of recalcitrant substrates oxidations is steadily increasing, show some useful features such as their noticeable operational stability along the catalytic cycle, and high effectiveness as oxidants for their hypervalent oxygenated derivatives (Figure 5). In fact, a nitrido-diiron-phthalocyanine in its resting state is a mixed-valence compound showing a formal oxidation number of +3.5, PcFe(III)–N=Fe(IV)Pc ↔ µ PcFe(IV)=N–Fe(III)Pc. Upon reaction with H2O2, a ultra-high valent peroxo species arises: O=Fe(IV)Pc–N=Fe(IV)Pc ↔ µ O=Fe(IV)Pc+•–N=Fe(IV)Pc, passing through a peroxo complex, formally analogous to Compound 0, such as the ultra-high valent complex is the formal analogue of Compound I [41]. With time, the unprecedented, exceptional oxidizing catalytic power of μ‑nitrido-bridged diiron complexes has been further explored, and such compounds are among the most promising oxidation and oxygenation catalysts for many practical applications, such as organic synthetic chemistry and degradation of recalcitrant pollutants. Outstanding examples are the controlled oxidation of methane at nearly ambient temperature and neutral pH [46] and the oxidative hydroxylation and dehalogenation of perfluoroaromatics [47], for which the reaction mechanism has been elucidated [48].
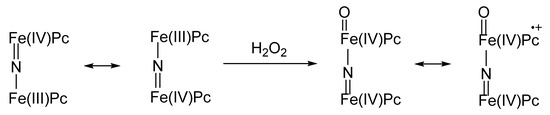
Figure 5. Nitrido-diiron-phthalocyanine with mixed valence state and ultra-high valent peroxo species formed after reaction with oxygen peroxide.
References
- Braun, A.; Tcherniac, J. Über die Produkte der Einwirkung von Acetanhydrid auf Phthalamid. Ber. Dtsch. Chem. Ges. 1907, 40, 2709–2714.
- Linstead, R.P.; Lowe, A.R. Phthalocyanines part III Preliminary experiments on the preparation of phthalocyanines from phthalonitrile. J. Chem. Soc. 1934, 1022–1027.
- Robertson, J.M.; Woodward, I. 37. An X-ray study of the phthalocyanines. Part III. Quantitative structure determination of nickel phthalocyanine. J. Chem. Soc. 1937, 10, 219–230.
- Moser, F.H.; Thomas, A.L. Phthalocyanine compounds. J. Chem. Educ. 1964, 41, 245.
- Claessens, C.G.; Hahn, U.; Torres, T. Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 2008, 8, 75–97.
- Gobo, N.R.S.; Brocksom, T.J.; de Oliveira, K.T. Soluble and Non-Aggregated Phthalocyanines: Synthesis, Mechanistic Aspects and Their Main Building Blocks. Curr. Org. Synth. 2017, 14, 1132–1155.
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191.
- Ferraudi, G.; Lappin, A.G. Review: Properties chemical reactivity of metallo phthalocyanine and tetramethylbenzoannulene complexes grafted into a polymer. J. Coord. Chem. 2014, 67, 3822–3839.
- Stillman, M.J.; Nyokong, T. Absorption and Magnetic Circular Dichroism Spectral Properties of Phthalocyanines Part 1: Complexes of the Dianion Pc(–2). In Phthalocyanines-Properties and Applications; Leznoff, C.C., Lever, A.B.P., Eds.; VCH Publishers, Inc.: New York, NY, USA, 1989; Volume 1, pp. 133–290.
- Milaeva, E.R.; Speier, G.; Lever, A.B.P. The Redox Chemistry of Metallophthalocyanines in Solution; Wiley-VCH: New York, NY, USA, 1993; Volume 3, pp. 3–69.
- Silva, A.M.S.; Neves, M.G.P.M.S.; Martins, R.R.L.; Cavaleiro, J.A.S.; Boschi, T.; Tagliatesta, P. Photo-oxygenation of meso-tetraphenylporphyrin derivatives: The influence of the substitution pattern and characterization of the reaction products. J. Porphyr. Phthalocyanines 1998, 2, 45–51.
- D’Alessandro, N.; Tonucci, L.; Dragani, L.K.; Morvillo, A.; Bressan, M. Fate of nickel and cobalt sulfophthalocyanines under oxidizing conditions: A spectroscopic investigation. J. Porphyr. Phthalocyanines 2003, 7, 484–492.
- D’Alessandro, N.; Tonucci, L.; Morvillo, A.; Dragani, L.K.; Di Deo, M.; Bressan, A. Thermal stability and photostability of water solutions of sulfophthalocyanines of Ru(II), Cu(II), Ni(II), Fe(III) and Co(II). J. Organomet. Chem. 2005, 690, 2133–2141.
- Jones, C.W. On the Stability and Recyclability of Supported Metal-Ligand Complex Catalysts: Myths, Misconceptions and Critical Research Needs. Top. Catal. 2010, 53, 942–952.
- Bressan, M.; Celli, N.; d’Alessandro, N.; Liberatore, L.; Morvillo, A.; Tonucci, L. Ruthenium sulfophthalocyanine catalyst for the oxidation of chlorinated olefins with hydrogen peroxide. J. Organomet. Chem. 2000, 593, 416–420.
- Nemykin, V.N.; Lukyanets, E.A. Synthesis of substituted phthalocyanines. Arkivoc 2010, 1, 136–208.
- Nyokong, T.; Gasyna, Z.; Stillman, M.J. Phthalocyanine. pi.-cation-radical species: Photochemical and electrochemical preparation of [ZnPc(-1).+ in solution. Inorg. Chem. 1987, 26, 548–553.
- Wang, G.; Ramesh, N.; Hsu, A.; Chu, D.; Chen, R. Density functional theory study of the adsorption of oxygen molecule on iron phthalocyanine and cobalt phthalocyanine. Mol. Simul. 2008, 34, 1051–1056.
- Shi, Z.; Zhang, J.J. Density functional theory study of transitional metal macrocyclic complexes’ dioxygen-binding abilities and their catalytic activities toward oxygen reduction reaction. J. Phys. Chem. C 2007, 111, 7084–7090.
- Dolotova, O.V.; Bundina, N.I.; Kaliya, O.L.; Lukyanets, E.A. Manganese Phthalocyanine Coordination Chemistry: Recent Results and Present Status. J. Porphyr. Phthalocyanines 1997, 1, 355–366.
- Wariishi, H.; Gold, M.H. Lignin peroxidase compound III. Mechanism of formation and decomposition. J. Biol. Chem. 1990, 265, 2070–2077.
- Lever, A.B.P.; Wilshire, J.P.; Quan, S.K. Oxidation of manganese(II) phthalocyanine by molecular-oxygen. Inorg. Chem. 1981, 20, 761–768.
- Uchida, K.; Soma, M.; Naito, S.; Onishi, T.; Tamaru, K. Manganese phthalocyanine as a model of tryptophan-2,3-dioxygenase. Chem. Lett. 1978, 7, 471–474.
- Moxon, N.T.; Fielding, P.E.; Gregson, A.K. Manganese 4,4’,4″,4′″-tetrasulfonated phthalocyanine, its dioxygen adduct, and electron-spin-resonance evidence for intramolecular electron-transfer in solution. J. Chem. Soc. Chem. Commun. 1981, 98–99.
- Ercolani, C.; Gardini, M.; Pennesi, G.; Rossi, G. Dioxygen Activation and Catalytic Oxidation of triphenylphosphine by iron phthalocyanine compounds. J. Mol. Catal. 1985, 30, 135–144.
- Kontarinis, D.; Paraskevas, S.M.; Paraskevas, M.S. Oxygen adducts of Co-phthalocyanine. Synth. React. Inorg. Met. Org. Chem. 2003, 33, 1381–1389.
- Hassanein, M.; Abdo, M.; Gerges, S.; El-Khalafy, S. Study of the oxidation of 2-aminophenol by molecular oxygen catalyzed by cobalt(II) phthalocyaninetetrasodiumsulfonate in water. J. Mol. Catal. A Chem. 2008, 287, 53–56.
- Schutten, J.H.; Piet, P.; German, A.L. Some observations on complexes of a cobalt phthalocyanine with poly(vinylamine) and their catalytic activity in the autoxidation of thiols. Die Makromol. Chem. 1979, 180, 2341–2350.
- Pan, Y.; Chen, W.X.; Lu, S.F.; Zhang, Y.F. Novel aqueous soluble cobalt phthalocyanine: Synthesis and catalytic activity on oxidation of 2-mercaptoethanol. Dye. Pigment. 2005, 66, 115–121.
- Geraskin, I.M.; Luedtke, M.W.; Neu, H.M.; Nemykin, V.N.; Zhdankin, V.V. Organic iodine(V) compounds as terminal oxidants in iron(III) phthalocyanine catalyzed oxidation of alcohols. Tetrahedron Lett. 2008, 49, 7410–7412.
- Kivits, P.; Debont, R.; Vanderveen, J. Vanadyl phthalocyanine: An organic material for optical data recording. Appl. Phys. A 1981, 26, 101–105.
- Ramadan, A.J.; Rochford, L.A.; Moffat, J.; Mulcahy, C.; Ryan, M.P.; Jones, T.S.; Heutz, S. The morphology and structure of vanadyl phthalocyanine thin films on lithium niobate single crystals. J. Mater. Chem. C 2016, 4, 348–351.
- Wong, E.W.Y.; Walsby, C.J.; Storr, T.; Leznoff, D.B. Phthalocyanine as a Chemically Inert, Redox-Active Ligand: Structural and Electronic Properties of a Nb(IV)-Oxo Complex Incorporating a Highly Reduced Phthalocyanine(4-) Anion. Inorg. Chem. 2010, 49, 3343–3350.
- Harris, D.L. High-valent intermediates of heme proteins and model compounds. Curr. Opin. Chem. Biol. 2001, 5, 724–735.
- Dolphin, D. The Electronic Configurations of Catalases and Peroxidases in their High Oxidation States: A Definitive Assessment. Isr. J. Chem. 1981, 21, 67–71.
- Jiang, G.; Chen, J.; Thu, H.Y.; Huang, J.S.; Zhu, N.; Che, C.M. Ruthenium porphyrin-catalyzed aerobic oxidation of terminal aryl alkenes to aldehydes by a tandem epoxidation-isomerization pathway. Angew. Chem. Int. Ed. 2008, 47, 6638–6642.
- Collman, J.P.; Barnes, C.E.; Brothers, P.J.; Collins, T.J.; Ozawa, T.; Gallucci, J.C.; Ibers, J.A. Oxidation of ruthenium(II) and ruthenium(III) porphyrins. Crystal structures of .mu.-oxo-bis[(p-methylphenoxo)(meso-tetraphenylporphyrinato)ruthenium(IV)] and ethoxo(meso-tetraphenylporphyrinato)(ethanol)ruthenium(III)-bisethanol. J. Am. Chem. Soc. 1984, 106, 5151–5163.
- Baek, H.K.; Vanwart, H.E. Elementary Steps in the Formation of Horseradish Peroxidase Compound I: Direct Observation of Compound 0, a New Intermediate with a Hyperporphyrin Spectrum. Biochemistry 1989, 28, 5714–5719.
- Zucca, P.; Neves, C.M.B.; Simoes, M.M.Q.; Neves, M.G.P.M.S.; Cocco, G.; Sanjust, E. Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes. Molecules 2016, 21, 964.
- Meunier, B.; Sorokin, A. Oxidation of pollutants catalyzed by metallophthalocyanines. Acc. Chem. Res. 1997, 30, 470–476.
- Sorokin, A.B.; Kudrik, E.V. Phthalocyanine metal complexes: Versatile catalysts for selective oxidation and bleaching. Catal. Today 2011, 159, 37–46.
- Sorokin, A.; Meunier, B. Oxidative Degradation of Polychlorinated Phenols Catalyzed by Metallosulfophthalocyanines. Chem. Eur. J. 1996, 2, 1308–1317.
- Floris, B.; Donzello, M.P.; Ercolani, C. Single-atom bridged dinuclear metal complexes with emphasis on phthalocyanine systems. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2003; pp. 1–62.
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160.
- Sorokin, A.B.; Tuel, A. Metallophthalocyanine functionalized silicas: Catalysts for the selective oxidation of aromatic compounds. Catal. Today 2000, 57, 45–59.
- Sorokin, A.B.; Kudrik, E.V.; Bouchu, D. Bio-inspired oxidation of methane in water catalyzed by N-bridged diiron phthalocyanine complex. Chem. Comm. 2008.
- Colomban, C.; Kudrik, E.V.; Afanasiev, P.; Sorokin, A.B. Degradation of chlorinated phenols in water in the presence of H2O2 and water-soluble mu-nitrido diiron phthalocyanine. Catal. Today 2014, 235, 14–19.
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of Oxidative Activation of Fluorinated Aromatic Compounds by N-Bridged Diiron-Phthalocyanine: What Determines the Reactivity? Chem. Eur. J. 2019, 25, 14320–14331.

