 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Heather Wilson | + 2838 word(s) | 2838 | 2021-01-12 11:54:04 | | | |
| 2 | Peter Tang | -96 word(s) | 2742 | 2021-02-21 13:10:23 | | |
Video Upload Options
There is a need to disentangle the etiological puzzle of age-related neurodegenerative diseases, whose clinical phenotypes arise from known, and as yet unknown, pathways that can act distinctly or in concert. Enhanced sub-phenotyping and the identification of in vivo biomarker-driven signature profiles could improve the stratification of patients into clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm. The rapidly growing field of neuroimaging offers valuable tools to investigate disease pathophysiology and molecular pathways in humans, with the potential to capture the whole disease course starting from preclinical stages. Positron emission tomography (PET) combines the advantages of a versatile imaging technique with the ability to quantify, to nanomolar sensitivity, molecular targets in vivo. There is an increasing body of literature implicating dysfunction of mitochondria and endoplasmic reticulum dynamics, energy metabolism and oxidative stress within the molecular paradigm of age-related neurodegenerative diseases. The development of novel PET radioligands enables the in vivo investigation of mitochondrial and ER dysfunction in age-related neurodegenerative diseases.
1. Introduction
It is widely accepted that age-related neurodegenerative diseases are increasingly becoming a global public health concern—in particular, Alzheimer’s disease (AD) and other late-onset dementias (LOD), with widespread socioeconomic and healthcare impacts worldwide. The increasing burden of age-related diseases is mainly due to the ageing world population and the unprecedented shift in aging demographics of individuals over 60 years of age, which is predicted to rise to two billion in 2050 [1]. Age-related neurodegenerative diseases encompass a spectrum of complex and heterogenous diseases, including AD, Parkinson’s disease (PD), Parkinson’s disease Dementia (PDD), Dementia with Lewy Bodies (DLB), the recently identified dementia form of “Limbic-predominant Age-related TDP-43 Encephalopathy (LATE)”, late-onset forms of Fronto-Temporal Dementia (FTD) and of Amyotrophic Lateral Sclerosis (ALS), as well as parkinsonian plus syndromes, such as Corticobasal Syndrome (CBS), Progressive Supranuclear Palsy (PSP) and Multiple System Atrophy (MSA). Unlike other public health challenges, such as cancer, which have seen the recent development of effective disease modifying treatments, therapies for age-related neurodegenerative diseases remain ineffective to modify the disease course, with most therapies only providing some symptomatic relief.
The majority of research across age-related neurodegenerative diseases is built upon the clinicopathological nosology model [2], whereby a specific clinical phenotype is studied aiming to unlock the underlying pathology, traditionally through post-mortem investigations and, more recently, through in vivo studies, using imaging and other biomarkers that reflect key pathological changes. Variations across diseases have been attributed to the selective vulnerability of specific neuronal subtypes to disease pathology that subsequently determine the clinical phenotypic expression. However, the majority of age-related neurodegenerative diseases are complex in nature, resulting from poorly understood interactions between genomic, environmental and lifestyle factors, across the life course, and harbor multiple pathologies; as a result, their clinical presentations can have distinct, as well as overlapping, features occurring at different levels and timepoints [3][4][5]. The concept that neuronal networks, rather than neuronal subtypes, could underlie differences in the clinical phenotype and the susceptibility of individuals to different neurodegenerative diseases has gained increasing interest over the last decade, aiming to unlock the paradigm of age-related neurodegenerative diseases [4][6][7][8][9]. In 2018, the National Institute on Aging and the Alzheimer’s Association proposed the Research Framework classification of AD to better define the diagnosis of AD, across a disease continuum from preclinical to severe clinical stages from other LOD forms, based on the in vivo AT(N) biomarker signature, corresponding to the three landmark pathological features of increased Amyloid and Tau burden, associated with a significant loss of volume and neurodegeneration [10][11]. While this classification is a step towards the application of biological signature profiles, consideration should be given to the use of a binary classification model for continuous variables, based on a predetermined threshold, as a predictive or diagnostic tool in clinical trials, especially when using a clinical phenotype, such as cognitive decline or dementia, as the primary outcome measure [12].
Almost all age-related neurodegenerative diseases can be classified into sporadic or familial forms. The discovery of fully penetrant genetic mutations in several familial neurodegenerative diseases has allowed for the investigation of the early disease pathology prior to the clinical manifestations, in these familial forms, prior to the manifestation of clinical symptoms that could help to unlock causal pathways. In the nonfamilial sporadic forms, in addition to the genetic variants that are noncausative but can confer susceptibility to disease, there is a wide range of risk factors that may affect disease onset and development, including environmental factors and exposures across the life course, cardiovascular status and hypertension, obesity, diabetes, sleep disorders and a variety of factors related to brain biological aging, such as protein misfolding and aggregation, epigenetics and perturbations in DNA damage and repair. However, not all patients with all, or some, of these risk factors will develop symptoms and signs amounting to a clinical diagnosis within their lifetime. While the interaction between genetics and disease mechanisms is indeed complex and has not been fully elucidated, it has been postulated that unraveling the genetics of age-related neurodegenerative diseases might form the basis for sub-phenotyping and/or reclassification based on genotypic divergence aiming to drive forward the application of precision medicine [13].
There is a need to disentangle the etiological puzzle of age-related neurodegenerative diseases, whose clinical phenotypes arise from known, and as yet unknown, pathways that can act distinctly or in concert (Figure 1). Over the last 40 years, preclinical animal studies and post-mortem evaluations have unlocked a number of disease mechanisms and therapeutic targets, which showed promise to translate into novel therapies for age-related neurodegenerative diseases. In AD, the main causal hypotheses involved the amyloid cascade and the tau phosphorylation-propagation hypothesis. However, the majority of clinical trials targeting these mechanisms have failed to meet their primary endpoints [14][15][16]. The failure of clinical trials, across age-related neurodegenerative diseases, could be due to a number of reasons such as the late initiation of treatments in the disease course, poor target engagement or selection of the tested compound, suboptimal cohort stratification and the inability to reach the required effect sizes due to inadequate sample size and/or short follow-up periods [15]. Moreover, an inadequate appreciation of the complexity of disease etiology and pathophysiology can lead to an oversimplified mono-therapeutic approach [16]
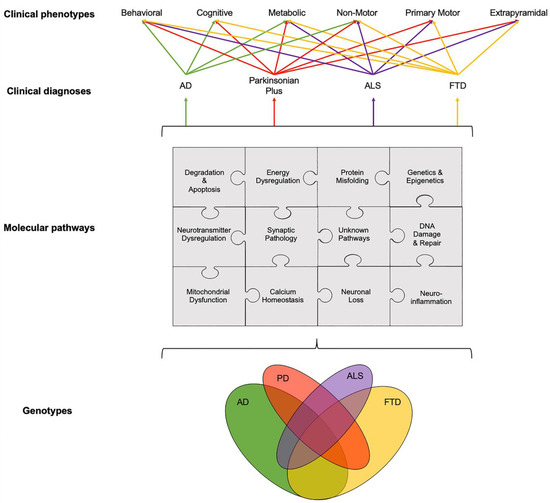
Figure 1. Schematic illustration of interlinked genotypes, molecular pathways and clinical phenotypes across age-related neurodegenerative diseases showing the overlap between various components and pathways at different levels, from genetics and molecular pathways to clinical phenotypes. Disentangling this etiological puzzle of known and yet unknown pathways acting distinctly or in concert could improve the stratification of patients into clinical trials and, potentially, help to drive the treatment landscape towards the precision medicine paradigm. The relationship between clinical diagnosis and clinical phenotypes was adapted from Ahmed et al., 2016 [3]. Abbreviations: AD: Alzheimer’s disease, ALS: Amyotrophic Lateral Sclerosis, FTD: Fronto-Temporal Dementia and PD: Parkinson’s disease.
While preclinical and post-mortem studies have, and will likely continue to, play a key role in the drug discovery process, as well as in understanding the underlying molecular mechanisms, considerations have to be given to their direct translation into humans [14][17]. For example, animal models of late-onset neurodegenerative diseases typically develop symptoms and die young, whereas in humans these diseases typically occur in late life [18]. Furthermore, post-mortem studies provide insights into pathological changes at a single timepoint (the very end stage of the disease), which can be contaminated from chronic drug treatments and other pathologies, making it difficult to disentangle whether the changes observed are a cause or consequence of neuronal death. The rapidly growing field of neuroimaging offers valuable tools to investigate disease pathophysiology and molecular pathways in vivo in humans, with the potential to capture the whole disease course. Positron emission tomography (PET) imaging combines the advantages of a versatile imaging technique with the ability to quantify, to nanomolar sensitivity, molecular targets, both in animals and in living humans. Magnetic resonance imaging (MRI) techniques can offer high spatial resolution and anatomical granularity with advanced acquisition protocols and analysis methodologies offering a platform to explore microstructural and functional connectivity, iron deposition, neuromelanin levels and neuro-hydrodynamics. Therefore, PET and MRI techniques are commonly employed in unison to extrapolate meaningful outcome measures reflecting molecular biology in vivo.
2. Dysregulation of Interlinked Molecular Pathways across Age-Related Neurodegenerative Diseases
While the temporal onset and the rate of progression can vary, clinical phenotypes, such as behavioral, cognitive, metabolic, nonmotor, primary motor and extrapyramidal, often overlap across different age-related neurodegenerative diseases (Figure 1). For example, patients with FTD can present with extrapyramidal symptoms similar to PD; AD patients can experience nonmotor symptoms such as sleep problems, which overlap with nonmotor symptoms observed in PD and parkinsonian plus syndromes, and patients with ALS can present with behavioral symptoms, such as apathy, which can overlap with FTD, parkinsonism plus syndromes and AD [19][20][21]. The pathogenesis and progression of age-related neurodegenerative diseases likely involves a dynamic interaction between various components and pathways at the genetic and pathological levels (Figure 1). Specific PET radioligands have been developed to target some of these molecular components, enabling the exploration of these pathways in vivo. There are a number of genotypic and molecular pathways that show varying degrees of overlap and crossover at various stages of disease etiology and progression. For example, while the clinical phenotype of three causative genes for FTD, C9orf72, MAPT and GRN, are associated with a similar behavioral variant FTD (bvFTD) presentation, the underlying protein pathology varies such that MAPT mutations are associated with tau pathology and C9orf72 and GRN mutations are associated with Tar-DNA-binding protein (TDP)-43 pathology [3]. Furthermore, a number of studies have unlocked genetic signatures that are common across different age-related neurodegenerative diseases. A meta-analysis of 1270 post-mortem brain tissue samples from AD, PD, ALS and Huntington’s disease (HD) patients identified shared gene expression signatures for 243 genes [22]. The common genes identified across these different diseases were related to functional pathways, including inflammation, synaptic signaling, metabolic dysfunction and oxidative stress. Moreover, while the causal role of epigenetics on age-related neurodegenerative diseases remains a topic of debate [23], similarities in the dysregulation of transcriptional networks and protein interaction networks have been reported [5].
It remains to be elucidated why, and how, pathologies diverge towards different clinical phenotypes and if there is a common causal mechanism that links the spectrum of age-related neurodegenerative diseases. The molecular nexopathies paradigm, introduced by Warren and colleagues, proposes that specific pathogenic proteins result in the disintegration of specific neural networks and multiple functional networks, which could give rise to phenotypic variations, as well as overlap between neurodegenerative diseases [4]. A deeper understanding of interlinked and distinctive molecular pathways, which drive pathological and clinical consequences, could provide novel therapeutic strategies. Molecular PET imaging can be employed to investigate known overlapping, and distinct, molecular pathologies and pathways in age-related neurodegenerative diseases (Figure 2). This section highlights novel PET biomarkers targeting mitochondrial dysfunction.
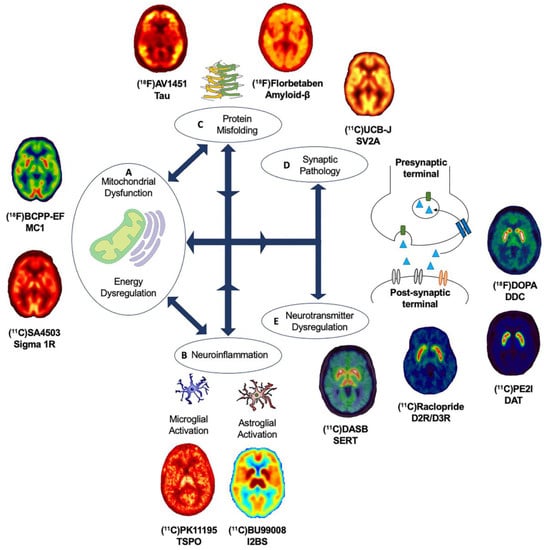
Figure 2. Overview of molecular pathways targeted with PET radioligands. (A) Mitochondrial dysfunction and energy dysregulation can be investigated using (18F)BCPP-EF, for mitochondrial complex 1 and (11C)SA4503 for sigma 1 receptor. (B) Neuroinflammation can be investigated by targeting translator protein expressed in the outer mitochondrial membrane and elevated in activated microglia using PET radioligands such as (11C)PK11195 and astroglia activation using novel PET radioligands such as (11C)BU99008 for imidazoline 2-binding sites expressed in the outer mitochondrial membrane. (C) Abnormal protein aggregation of tau and amyloid-β can be quantified using specific radioligands such as (18F)AV1451 and (18F)Florbetaben, respectively. (D) Synaptic pathology can be investigated using (11C)UCB-J targeting synaptic vesicle glycoprotein 2A. (E) Dysregulation of neurotransmitter systems can be investigated by employing various PET radioligands, including serotonergic markers such as (11C)DASB for the serotonin transporter and dopaminergic markers such as presynaptic markers (18F)DOPA for dopamine storage, (11C)PE2I for dopamine transporter and (11C)Raclopride for postsynaptic dopaminergic receptors, as well as PET radioligands for noradrenergic, glutamatergic and GABAergic systems. Abbreviations: D2R/D3R: Dopamine type-2/type-3 receptor, DAT: Dopamine transporter, DDC: Dopa Decarboxylase, I2BS: Imidazoline 2-binding sites, MC1: Mitochondrial Complex 1, SERT: Serotonin transporter, Sigma 1R: Sigma 1 receptor, SV2A: Synaptic vesicle glycoprotein 2A and TSPO: Translocator protein.
3. Mitochondrial Dysfunction and Energy Dysregulation
There is an increasing body of literature implicating dysfunction of mitochondria and endoplasmic reticulum (ER) dynamics, energy metabolism and oxidative stress within the molecular paradigm of age-related neurodegenerative diseases [24][25][26][27][28][29]. Protein aggregation and deposition have been linked with mitochondrial dysfunction, disrupted mitochondrial transport, dysregulation of adenosine triphosphate (ATP) production, calcium imbalance and oxidative stress [28]. Furthermore, mitochondrial dysfunction can alter the energy supply to synapses, which could drive synaptic disconnection, contributing towards synaptic dysfunction and loss [30][31]. The identification of several genes, such as PINK-1, Parkin, TREM2, APOE and TOMM40 [32][33][34][35], which play key roles in the normal functioning of mitochondria has also highlighted the role of mitochondrial dysfunction in disease pathogenesis [36][37][38]. The temporal sequence of events and the exact interplay between mitochondria and ER dysfunction, oxidative stress, neuroinflammation and protein deposition remains to be fully elucidated. There are lines of evidence to support the accumulation of toxic proteins preceding and triggering mitochondrial and ER dysfunction [39][40][41]. Conversely, other evidence suggests that mitochondrial dysfunction and, consequently, oxidative stress and calcium imbalance, together with dysfunction of the ER, may lead to protein misfolding and the accumulation of toxic protein aggregates [42][43].
The development of novel PET radioligands, (18F)BCPP-EF, for mitochondrial complex 1 (MC1) and (11C)SA-4503 for sigma 1 receptor (σ1R) enables the in vivo investigation of mitochondrial and ER dysfunction (Figure 2A) in late-onset neurodegenerative and other diseases related to aging [44][45]. Sigma-1 receptors are expressed at the mitochondrion-associated ER membrane, where they regulate calcium signaling from the ER to the mitochondrion [46][47][48]. Sigma-1 receptors also display neuromodulator and neuroprotective properties, aiding protein folding and modulating synaptic neurotransmitter functions [46][49][50]. MC1 plays a fundamental role in cellular energy production, acting as the first rate limiting step of oxidative phosphorylation in the electron transport chain in mitochondria, as well as maintaining calcium homeostasis and regulating reactive oxygen species (ROS) levels [51][52]. The altered expression and dysfunction of σ1R and MC1 have been illustrated from post-mortem and preclinical studies in ALS, AD and PD [49][50][53][54][55][56][57].
Recently, σ1R and MC1 levels were investigated in a cohort of early de novo PD patients using (11C)SA-4503 and (18F)BCPP-EF PET, respectively [58]. Lower levels of σ1R and MC1 were observed at the baseline, but there were no significant cross sectional or longitudinal changes at 12-months follow-up. In another small cohort of moderate levodopa-treated PD patients, decreased striatal σ1R levels was reported [59][60], suggesting that the loss of σ1R might be more prominent in moderate-to-advanced disease stages. A combined (18F)BCPP-EF and (11C)PE2I PET preclinical study demonstrated that the striatal loss of MC1 correlated with the loss of presynaptic nigrostriatal dopaminergic neurons, supporting the interplay and colocalization of mitochondrial and synaptic dysfunction in a PD model [61]. Work is ongoing to investigate the role of σ1R and MC1 in AD, ALS, FTD and HD using (11C)SA-4503 and (18F)BCPP-EF PET, respectively, as part of the MIND-MAPS program (https://lp.invicro.com/mind-maps), which could help to provide a more comprehensive understanding of the mitochondrial-ER-synaptic complex, across the spectrum of age-related neurodegenerative diseases. Preliminary work suggests decreased MC1 density in AD [62] and FTD patients [63], with the loss of MC1 associated with global cognitive impairment across cohorts of age-related neurodegenerative cohorts [64]. Furthermore, preliminary findings indicate that σ1R density is increased in early AD, suggesting that this may represent a potential cellular response to stress that could subsequently decrease as the disease progresses [62]. Reduced (18F)BCPP-EF uptake has also been shown to correlate with increase tau deposition, using (11C)PPB3 PET, but not with amyloid-β, using (11C)PiB PET, or glucose metabolism, using (18F)FDG PET [65]. These preliminary findings could indicate that tau pathology precedes early mitochondria-related energy failure. However, these findings need to be further validated in larger, longitudinal studies. The temporal relationship between mitochondrial dysfunction, energy dysregulation and synaptic neuropathology warrants further investigation, as it may play a key role in the development of several age-related neurodegenerative diseases.
There are a number of other PET radioligands with mitochondrial targets. First-, second- and third-generation PET radioligands, such as (11C)PK11195, (11C)PBR28, and (11C)ER176 respectively, commonly employed to study neuroinflammation target the 18-kDa translator protein (TSPO) expressed in the outer mitochondrial membrane and elevated in activated microglia. The novel PET radioligand (11C)BU99008 targets imidazoline 2-binding sites expressed in the outer mitochondrial membrane of activated astrocytes. Furthermore, PET radioligands such as (11C)Harmine and (11C)Deprenyl target mitochondrial monoamine oxidase (MAO)-A and MAO-B, respectively.
References
- United Nations, D.o.E.a.S.A., Population Division. World Population Ageing; United Nations: New York, NY, USA, 2013.
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol 2017, 13, 457-476, doi:10.1038/nrneurol.2017.96.
- Ahmed, R.M.; Devenney, E.M.; Irish, M.; Ittner, A.; Naismith, S.; Ittner, L.M.; Rohrer, J.D.; Halliday, G.M.; Eisen, A.; Hodges, J.R., et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2016, 87, 1234-1241, doi:10.1136/jnnp-2014-308350.
- Warren, J.D.; Rohrer, J.D.; Schott, J.M.; Fox, N.C.; Hardy, J.; Rossor, M.N. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci 2013, 36, 561-569, doi:10.1016/j.tins.2013.06.007.
- Arneson, D.; Zhang, Y.; Yang, X.; Narayanan, M. Shared mechanisms among neurodegenerative diseases: from genetic factors to gene networks. J Genet 2018, 97, 795-806.
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Dissecting the Molecular Mechanisms of Neurodegenerative Diseases through Network Biology. Front Aging Neurosci 2017, 9, 166, doi:10.3389/fnagi.2017.00166.
- Pievani, M.; de Haan, W.; Wu, T.; Seeley, W.W.; Frisoni, G.B. Functional network disruption in the degenerative dementias. Lancet Neurol 2011, 10, 829-843, doi:10.1016/S1474-4422(11)70158-2.
- Eisen, A.; Turner, M.R. Does variation in neurodegenerative disease susceptibility and phenotype reflect cerebral differences at the network level? Amyotroph Lateral Scler Frontotemporal Degener 2013, 14, 487-493, doi:10.3109/21678421.2013.812660.
- Chhatwal, J.P.; Schultz, A.P.; Johnson, K.A.; Hedden, T.; Jaimes, S.; Benzinger, T.L.S.; Jack, C., Jr.; Ances, B.M.; Ringman, J.M.; Marcus, D.S., et al. Preferential degradation of cognitive networks differentiates Alzheimer's disease from ageing. Brain 2018, 141, 1486-1500, doi:10.1093/brain/awy053.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J., et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018, 14, 535-562, doi:10.1016/j.jalz.2018.02.018.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S., et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539-547, doi:10.1212/WNL.0000000000002923.
- McRae-McKee, K.; Udeh-Momoh, C.T.; Price, G.; Bajaj, S.; de Jager, C.A.; Scott, D.; Hadjichrysanthou, C.; McNaughton, E.; Bracoud, L.; Ahmadi-Abhari, S., et al. Perspective: Clinical relevance of the dichotomous classification of Alzheimer's disease biomarkers: Should there be a "gray zone"? Alzheimers Dement 2019, 15, 1348-1356, doi:10.1016/j.jalz.2019.07.010.
- Sturchio, A.; Marsili, L.; Mahajan, A.; Grimberg, M.B.; Kauffman, M.A.; Espay, A.J. How have advances in genetic technology modified movement disorder nosology? Eur J Neurol 2020, 27, 1461-1470, doi:10.1111/ene.14294.
- Burns, T.C.; Verfaillie, C.M. From mice to mind: Strategies and progress in translating neuroregeneration. Eur J Pharmacol 2015, 759, 90-100, doi:10.1016/j.ejphar.2015.03.041.
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, doi:10.3390/biomedicines7040097.
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement 2016, 12, 60-64, doi:10.1016/j.jalz.2015.12.003.
- Burns, T.C.; Li, M.D.; Mehta, S.; Awad, A.J.; Morgan, A.A. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: A systematic bioinformatics-based critique of preclinical models. Eur J Pharmacol 2015, 759, 101-117, doi:10.1016/j.ejphar.2015.03.021.
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front Aging Neurosci 2015, 7, 168, doi:10.3389/fnagi.2015.00168.
- Kobylecki, C.; Jones, M.; Thompson, J.C.; Richardson, A.M.; Neary, D.; Mann, D.M.; Snowden, J.S.; Gerhard, A. Cognitive-behavioural features of progressive supranuclear palsy syndrome overlap with frontotemporal dementia. J Neurol 2015, 262, 916-922, doi:10.1007/s00415-015-7657-z.
- Mioshi, E.; Caga, J.; Lillo, P.; Hsieh, S.; Ramsey, E.; Devenney, E.; Hornberger, M.; Hodges, J.R.; Kiernan, M.C. Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 2014, 82, 149-155, doi:10.1212/WNL.0000000000000023.
- Borroni, B.; Alberici, A.; Agosti, C.; Cosseddu, M.; Padovani, A. Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int Psychogeriatr 2009, 21, 463-468, doi:10.1017/S1041610209008862.
- Li, M.D.; Burns, T.C.; Morgan, A.A.; Khatri, P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Commun 2014, 2, 93, doi:10.1186/s40478-014-0093-y.
- Millan, M.J. An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology 2013, 68, 2-82, doi:10.1016/j.neuropharm.2012.11.015.
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35-45, doi:10.1016/j.mito.2019.07.003.
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, doi:10.3390/cells9041065.
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front Pharmacol 2019, 10, 902, doi:10.3389/fphar.2019.00902.
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Frontiers in Neuroscience 2019, 13, doi:10.3389/fnins.2019.00560.
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 2012, 342, 619-630, doi:10.1124/jpet.112.192138.
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013, 12, 105-118, doi:10.1016/S1474-4422(12)70238-7.
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson's disease. Cold Spring Harb Perspect Med 2012, 2, a009332, doi:10.1101/cshperspect.a009332.
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis 2010, 20 Suppl 2, S499-512, doi:10.3233/JAD-2010-100504.
- Roses, A.; Sundseth, S.; Saunders, A.; Gottschalk, W.; Burns, D.; Lutz, M. Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer's disease. Alzheimers Dement 2016, 12, 687-694, doi:10.1016/j.jalz.2016.03.015.
- Prokopenko, I.; Miyakawa, G.; Zheng, B.; Heikkinen, J.; Petrova Quayle, D.; Udeh-Momoh, C.; Claringbould, A.; Neumann, J.; Haytural, H.; Kaakinen, M.A., et al. Alzheimer's disease pathology explains association between dementia with Lewy bodies and APOE-epsilon4/TOMM40 long poly-T repeat allele variants. Alzheimers Dement (N Y) 2019, 5, 814-824, doi:10.1016/j.trci.2019.08.005.
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci 2015, 40, 200-210, doi:10.1016/j.tibs.2015.02.003.
- Chiba-Falek, O.; Gottschalk, W.K.; Lutz, M.W. The effects of the TOMM40 poly-T alleles on Alzheimer's disease phenotypes. Alzheimers Dement 2018, 14, 692-698, doi:10.1016/j.jalz.2018.01.015.
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson's Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep 2018, 18, 21, doi:10.1007/s11910-018-0829-3.
- Gottschalk, W.K.; Lutz, M.W.; He, Y.T.; Saunders, A.M.; Burns, D.K.; Roses, A.D.; Chiba-Falek, O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. J Parkinsons Dis Alzheimers Dis 2014, 1, doi:10.13188/2376-922X.1000003.
- Ridge, P.G.; Kauwe, J.S.K. Mitochondria and Alzheimer's Disease: the Role of Mitochondrial Genetic Variation. Curr Genet Med Rep 2018, 6, 1-10, doi:10.1007/s40142-018-0132-2.
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfo, E.; Carmona, M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm 2011, 118, 821-839, doi:10.1007/s00702-010-0482-8.
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol 2017, 134, 129-149, doi:10.1007/s00401-017-1704-z.
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson's disease. Trends Neurosci 2015, 38, 178-188, doi:10.1016/j.tins.2014.12.009.
- Zaltieri, M.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A. Mitochondrial Dysfunction and alpha-Synuclein Synaptic Pathology in Parkinson's Disease: Who's on First? Parkinsons Dis 2015, 2015, 108029, doi:10.1155/2015/108029.
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A., et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem 2005, 92, 628-636, doi:10.1111/j.1471-4159.2004.02895.x.
- Mansur, A.; Rabiner, E.A.; Comley, R.A.; Lewis, Y.; Middleton, L.T.; Huiban, M.; Passchier, J.; Tsukada, H.; Gunn, R.N.; Consortium, M.-M. Characterization of 3 PET Tracers for Quantification of Mitochondrial and Synaptic Function in Healthy Human Brain: (18)F-BCPP-EF, (11)C-SA-4503, and (11)C-UCB-J. J Nucl Med 2020, 61, 96-103, doi:10.2967/jnumed.119.228080.
- Mansur, A.; Rabiner, E.A.; Tsukada, H.; Comley, R.A.; Lewis, Y.; Huiban, M.; Passchier, J.; Gunn, R.N. Test-retest variability and reference region-based quantification of (18)F-BCPP-EF for imaging mitochondrial complex I in the human brain. J Cereb Blood Flow Metab 2020, Epub ahead of print , doi:10.1177/0271678X20928149.
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596-610, doi:10.1016/j.cell.2007.08.036.
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: more than just a housekeeper. Trends Cell Biol 2009, 19, 81-88, doi:10.1016/j.tcb.2008.12.002.
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 2006, 174, 915-921, doi:10.1083/jcb.200604016.
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 2014, 137, 1998-2014, doi:10.1093/brain/awu107
- Tsai, S.Y.; Pokrass, M.J.; Klauer, N.R.; De Credico, N.E.; Su, T.P. Sigma-1 receptor chaperones in neurodegenerative and psychiatric disorders. Expert Opin Ther Targets 2014, 18, 1461-1476, doi:10.1517/14728222.2014.972939.
- Papa, S.; De Rasmo, D. Complex I deficiencies in neurological disorders. Trends Mol Med 2013, 19, 61-69, doi:10.1016/j.molmed.2012.11.005.
- Sazanov, L.A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol 2015, 16, 375-388, doi:10.1038/nrm3997.
- Mavlyutov, T.A.; Guo, L.W.; Epstein, M.L.; Ruoho, A.E. Role of the Sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J Pharmacol Sci 2015, 127, 10-16, doi:10.1016/j.jphs.2014.12.013.
- Jansen, K.L.; Faull, R.L.; Storey, P.; Leslie, R.A. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer's disease correlates with CA1 pyramidal cell loss. Brain Res 1993, 623, 299-302, doi:10.1016/0006-8993(93)91441-t.
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv Exp Med Biol 2017, 964, 133-152, doi:10.1007/978-3-319-50174-1_10.
- Flones, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.B.; Haugarvoll, K., et al. Neuronal complex I deficiency occurs throughout the Parkinson's disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol 2018, 135, 409-425, doi:10.1007/s00401-017-1794-7.
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990, 54, 823-827, doi:10.1111/j.1471-4159.1990.tb02325.x.
- Wilson, H.; Pagano, G.; de Natale, E.R.; Mansur, A.; Caminiti, S.P.; Polychronis, S.; Middleton, L.T.; Price, G.; Schmidt, K.F.; Gunn, R.N., et al. Mitochondrial Complex 1, Sigma 1, and Synaptic Vesicle 2A in Early Drug-Naive Parkinson's Disease. Mov Disord 2020, 35, 1416-1427, doi:10.1002/mds.28064.
- Toyohara, J.; Sakata, M.; Ishiwata, K. Imaging of sigma1 receptors in the human brain using PET and [11C]SA4503. Cent Nerv Syst Agents Med Chem 2009, 9, 190-196.
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M., et al. Function of sigma1 receptors in Parkinson's disease. Acta Neurol Scand 2005, 112, 103-107, doi:10.1111/j.1600-0404.2005.00432.x.
- Kanazawa, M.; Ohba, H.; Nishiyama, S.; Kakiuchi, T.; Tsukada, H. Effect of MPTP on Serotonergic Neuronal Systems and Mitochondrial Complex I Activity in the Living Brain: A PET Study on Conscious Rhesus Monkeys. J Nucl Med 2017, 58, 1111-1116, doi:10.2967/jnumed.116.189159.
- Venkataraman, A.; Mansur, A.; Lewis, Y.; Kocagoncu, E.; Lingford-Hughes, A.; Huiban, M.; Passchier, J.; Rowe, J.; Tsukada, H.; Brooks, D., et al. Evaluation of mitochondrial and synaptic and synaptic function in Alzheimer’s disease: a [18F]BCPP-EF, [11C]SA4503 and [11C]UCB-J PET study. In Proceedings of 29th International Symposium on Cerebral Blood Flow, Metabolism and Function. Yokohama, Japan, 4-7 July 2019, 39, 121-122.
- Clarke, M.; Mansur, A.; Passchier, J.; Lewis, Y.; Evans, K.; Chen, L.; Schwarz, A.; Takano, A.; Gunn, R.; Cash, D., et al. Imaging synaptic and mitochondrial function in frontotemporal dementia using [11C]UCB-J, [18F]BCPP-EF and [11C]SA4503 PET. In Proceedings of Human Amyloid Imaging, Miami, FL, USA, 15-17 January 2020.
- Rabiner, E.; Mansur, A.; Venkataraman, A.; Price, G.; Wilson, H.; Pagano, G.; Clarke, M.; Lewis, Y.; Matthews, P.M.; Rowe, J.B., et al. MIND MAPS: Assessment of the mitochondrial - endoplasmic reticulum - synaptic axis in neurodegeneration by [18F]BCPP-EF, [11C]SA4503 and [11C]UCB-J PET imaging. In Proceedings of Human Amyloid Imaging, Miami, FL, USA, 15-17 January 2020.
- Terada, T.; Therriault, J.; Su, P.K.M.; Savard, M.; Ouchi, Y.; Rosa-Neto, P. In vivo association of mitochondrial dysfunction with tau pathology in early Alzheimer’s disease. In Proceedings of Human Amyloid Imaging, Miami, FL, USA, 15-17 January 2020.

