 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shuihong Li | + 2350 word(s) | 2350 | 2021-02-04 05:10:17 | | | |
| 2 | Shuihong Li | -8 word(s) | 2342 | 2021-02-05 03:51:55 | | | | |
| 3 | Dean Liu | + 27 word(s) | 2369 | 2021-02-07 08:38:48 | | | | |
| 4 | Catherine Yang | Meta information modification | 2369 | 2021-09-28 11:25:06 | | |
Video Upload Options
Mycoplasma pneumoniae is a major causative agent of community-acquired pneumonia which can lead to both acute upper and lower respiratory tract inflammation, and extrapulmonary syndromes.
1. Introduction
Community-acquired pneumonia (CAP) is associated with high morbidity and mortality, and the disease is also a major threat to public health worldwide[1]. About 8–40% of CAP in children admitted to hospitals were caused by Mycoplasma pneumoniae[2][3][4]. Based on the reported cases in China, M. pneumoniae infections accounted for 19.2% of all CAP cases in adults, and the prevalence of CAP in children and teenagers, ranged from 10% to 30% [1][5]. In the USA, a recent study of 2254 hospitalized children with CAP showed that 8% children with median age of 7 years were positive for M. pneumoniae by polymerase chain reaction (PCR)[6].
Airborne droplets containing M. pneumoniae can be transmitted and spread among people through coughing and sneezing. M. pneumoniae causes both upper and lower respiratory tract infections, and in most cases the clinical symptoms are non-specific[7]. Tracheobronchitis is the most common type of lower respiratory infection, the incidence of which is about 20 times that of pneumonia, and 10–40% of respiratory tract infections caused by M. pneumoniae will eventually develop into pneumonia[8]. While most pneumonia caused by M. pneumoniae (MPP) cases are benign, some cases may develop into severe pneumonia and refractory pneumonia with pleural effusion, multi-organ dysfunction, and serious long-term sequelae, including bronchiolitis obliterans and bronchiectasis[9]. Although CAP is the most significant disease caused by M. pneumoniae, the pathogen is known to cause upper respiratory tract infections. Pharyngitis is commonly reported while rhinosinusitis and otitis media are less frequently encountered in upper respiratory tract infections caused by M. pneumoniae[7].
M. pneumoniae respiratory infections are associated with asthma exacerbation during which patients will suffer from a combination of symptoms including sudden or progressive coughing, respiratory distress, wheezing or chest pain [10][11]. The onset of asthma is due to the release of Mycoplasma-mediated cytokine in infected patients[12]. Respiratory infections caused by M. pneumoniae are also associated with a wide array of extrapulmonary manifestations such as meningoencephalitis, myocarditis, nephritis, atherosclerosis and mucocutaneous eruptions, etc.[13][14][15][16][17]. More importantly, M. pneumoniae induces mucocutaneous diseases include Stevens-Johnson syndrome and M. pneumoniae-associated mucositis. These mucocutaneous diseases are frequently associated with systemic inflammation and higher risk of the occurrence of long-term sequelae[18][19][20][21].
Due to the atypical symptoms produced during M. pneumoniae infection, pneumonia can be underestimated during the early stage of infection. There are no distinctive clinical or radiographic features in patients with M. pneumoniae infections, so laboratory diagnosis mainly based on rapid culture of throat swab specimens, PCR and serological assays. Furthermore, enzyme-linked immunosorbent assays (ELISA) detecting the N-terminal fragment of P116 protein and the C-terminal region of P1 protein both hold promise for serodiagnosis[22][23]. The IgM ELISA assays based on the short recombinant P116 and P1 proteins were shown to improve the specificity of the immunodiagnostic assay[22].
Although M. pneumoniae infection is generally self-limiting and does not require antibiotic treatment, patients of all age groups can develop severe, life-threatening or extrapulmonary diseases[24]. Antibiotics such as tetracycline and fluoroquinolone have been reported to be effective in eliminating M. pneumoniae infections[25] but tetracyclines cause discoloration of bones and teeth in young children. Fluoroquinolones can also affect the muscle, joint and tendon. Instead, macrolides, which have fewer side effects, have been the drug of choice for treating M. pneumoniae infection in past years[26]. More worrisome is that the extensive use of macrolides in China has led to a particularly high rate of macrolide resistance in this organism (69%~95%)[27]. The emergence of antibiotic resistance represents another challenge regarding the treatment of M. pneumoniae infections. Failure in antibiotic treatment has caused an increase in mortality rate during recent years[28]. Although the clinical outcomes of infections caused by macrolide-susceptible and -resistant M. pneumoniae isolates are not significantly different, patients infected with macrolide-resistant isolates had a longer febrile period (1.71 days), length of hospital stay (1.61 day), antibiotic drug courses (2.93 days), and defervescence time after macrolide treatment (2.04 days) compared to patients infected with macrolide-sensitive isolates[29]. Furthermore, macrolide-resistant strains may be associated with more extrapulmonary complications, and severe clinical and radiological features[24][30]. Hence, the development of vaccines against M. pneumoniae infections is a potential solution for the prevention of infections caused by the pathogen.
2. Virulence and Pathogenesis of M. pneumoniae
M. pneumoniae encodes a variety of virulence factors, which include adhesins, glycolipids, toxic metabolites, community-acquired respiratory distress syndrome (CARDS) toxin, and capsular polysaccharides. Table 1 summarizes the key virulence factors associated with M. pneumoniae.
Table 1. Key virulence factors of M. pneumoniae.
| Pathogenic Mechanism | Virulence Factor | Gene Annotation | Reference |
|---|---|---|---|
| Adherence | P1 | MPN141 | [31] |
| P30 | MPN453 | [32][33] | |
| P40 (Protein C) | MPN142 | [34] | |
| P90 (Protein B) | MPN142 | [34] | |
| P200 | MPN567 | [35] | |
| Hypothetical protein HMW1-3 (high molecular weight) | MPN447/310/452 | [36] | |
| P116 | MPN213 | [36] | |
| P65 | MPN309 | [37] | |
| Elongation factor thermo unstable (EF-Tu) | MPN665 | [38][39][40] | |
| Pyruvate dehydrogenase subunit B | MPN392 | [41] | |
| Glycolytic enzymes enolase | MPN606 | [41][42] | |
| TopJ | MPN119 | [43] | |
| Immune evasion | Nuclease | MPN491 | [44] |
| Immunoglobin binding protein (IbpM) | MPN400 | [45] | |
| Inflammation injury | H2O2 | / | [46] |
| Reactive oxygen species (ROS) | / | [46] | |
| H2S | / | [47] | |
| HapE enzyme | MPN487 | [47][48] | |
| Oxidase GlpO | MPN051 | [49] | |
| Membrane lipids | / | [50] | |
| Membrane lipoproteins | / | [51] | |
| Capsular materials | / | [52] | |
| Cytotoxicity | Community-Acquired Respiratory Distress Syndrome (CARDS) toxin | MPN372 | [53][54] |
| Cytotoxic nuclease | MPN133 | [55] | |
| Gliding motility | P65 | MPN309 | [37] |
| P30 | MPN453 | [32] | |
| Hypothetical protein MPN387 | MPN387 | [56] | |
| P24 | MPN312 | [57] | |
| P41 | MPN311 | [57] |
2.1. Adhesins
M. pneumoniae attaches to epithelial cell surfaces with a high affinity for human respiratory epithelial cells. The pathogen has no cell wall and colonizes the respiratory tract via its specific attachment organelle, which is a protrusion at one end of the Mycoplasma pneumoniae cell (Figure 1). The attachment organelle consists of internal and surface structures[58]. The internal structure is made up of a dumbbell-shaped terminal button consisting of three protein molecules (HMW2, HMW3, and P65), paired plates (HMW1, HMW2, CpsG, and HMW3), and a bowl complex (Lon, P24, TopJ, P200, P41, MPN387, and HMW2). The Nap structure in the surface adhesion complex consists of the main adhesins (P1 and P30) and accessory proteins (P40 and P90) surrounding the cell membrane (Figure 1). During gliding, the force generated at the bowl complexes is transmitted through the paired plates and reaches the P1 adhesin complex[58]. P30 adhesin is a membrane protein at the distal end of the attachment organelle, required for cytoadherence, gliding motility and stabilization of the accessory protein P65[33]. Interaction of the M. pneumoniae attachment organelle with the host’s respiratory epithelium induces cytoskeleton rearrangement in the host cell, which promotes intracellular delivery of the pathogen[59][60].
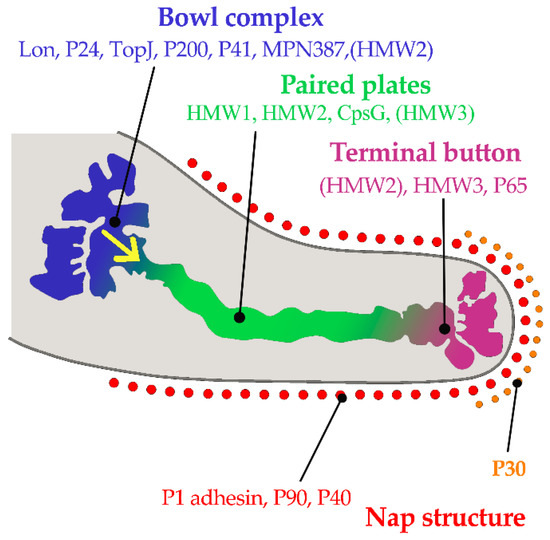
Figure 1. Component proteins of the internal structure of attachment organelle and proposed mechanism of movements for gliding in M. pneumoniae. HMW1, HMW2, and HMW3 refer to three high molecular weight (HMW) proteins. The force is generated at the bowl complexes, transmitted through the paired plates, and reaches the P1 adhesin complex in the direction of the yellow arrow. (Based on ideas from Nakane, et al.[58]). Copyright: ©2015. Public Library of Science. Creative Commons Attribution License and disclaimer available from: http://creativecommons.org/licenses/by/4.0/.
The host receptors for M. pneumoniae are sialylated glycoproteins on the respiratory epithelium. The nature and density of host receptor moieties affect the attachment and gliding mobility of the pathogen. P1 adhesin binds to both α-2,3 and α-2,6 linkages, but only the latter type of linkage supports gliding of M. pneumoniae[61].
Attachment and invasion of M. pneumoniae produces direct damage to the host’s respiratory epithelium[59][62]. Disturbance of carbohydrate metabolism, amino acid intake and protein synthesis of the host cell results in nutrient depletion[46] Furthermore, the oxygen radicals generated by the pathogen in the host cell can lead to cilia destruction and host cell damage[63][64].
2.2. Inflammation Injury
Bacterial cellular components, metabolites and toxins released from M. pneumoniae are able to induce damage in the host tissues. These include cytotoxicity, oxidative damage, apoptosis and immune-pathological damage.
2.2.1. Enzymes and Metabolites
The enzyme, HapE, of M. pneumoniae is a virulence factor that can produce H2S by the desulfurization of cysteine [47][48] which can lead to erythrocyte lysis. This enzyme mediates inflammatory reactions via adenosine triphosphate (ATP)-sensitive K+ channels[65]. Oxidation of glycerol by the pathogen produces toxic metabolites[66] including hydrogen peroxide [67][68] which injures cells by causing inflammation. In addition, the Ca2+-dependent cytotoxic nuclease (encoded by MPN133) produced by M. pneumoniae can lead to apoptotic-like programmed cell death in the host.
2.2.2. Lipoproteins
More than 50 different lipoproteins have been identified in M. pneumoniae, many of them involved in inflammatory reactions[69]. The transcription of M. pneumoniae lipoprotein genes are regulated in response to changes in environmental conditions (e.g., oxidative and acidic stress)[70][71]. The N-terminal region of all the lipoproteins contains a lipid-cysteine structure and these lipoproteins induce inflammation[69]. M. pneumoniae lipoproteins can be recognized by toll-like receptor (TLR)1, TLR2 and TLR6, which stimulate the release of proinflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and other inflammatory mediators via the nuclear factor κB (NF-κB) pathway[72][73].
2.2.3. Community-Acquired Respiratory Distress Syndrome (CARDS) Toxin
The CARDS toxin encoded by MPN372 is a unique bacterial adenosine diphosphate (ADP)-ribosylating and vacuolating toxin produced by M. pneumoniae[74][75]. The structure of CARDS toxin comprises a triangular molecule in which N-terminal mono-ADP ribosyl-transferase (mART) and C-terminal tandem β-trefoil domains associate to form a unique overall architecture different from other well-recognized ADP-ribosylating bacterial toxins [53]. CARDS toxin demonstrates high binding affinity to human surfactant protein A and annexin A2 when present in the airway epithelia and exhibits specific biological activities including mono-ADP ribosylation and vacuolization[53][74]. CARDS toxin binds to mammalian cell surface receptors and is internalized rapidly in a dose and time-dependent manner. The internalization process is mediated by clathrin molecules, which form a molecular scaffold for uptake of CARDS toxin[76]. The toxin is cytotoxic to mammalian cells by activation of the NLRP3-associated inflammasome and further promotes the release of IL-1β and IL-18[77][78][79]]. CARDS toxin increases the expression of the proinflammatory cytokines IL-1β, IL-6 and TNF-α in a dose- and activity-dependent manner [80]. CARDS toxin is capable of inducing an allergic-type inflammation in animals[82][81], but there is no convincing evidence that CARDS toxin is a causal factor of M. pneumoniae-associated asthma.
2.2.4. Lipids
The cell membrane of M. pneumoniae has a high lipid content (comprising primarily of the acidic glycerophospholipids phospholipids and cholesterol), which can infiltrate the host epithelial cells, disrupt the lipid bilayer of the cell membrane and cause leakage of ionic metabolites[54][55]. Furthermore, some scholars speculate that these lipids may act as potential TLR4 ligands for binding to TLR4 and elicit macrophage autophagy, eventually leading to the secretion of proinflammatory cytokines[50][83] and triggering typical host cell inflammatory responses [50][84].
2.2.5. Capsules
M. pneumoniae has a capsular structure made up of polysaccharides[52] which may be potential virulence factors and are immunogenic, but its functional role in pathogenesis remains unclear and needs to be further explored [85][86][87].
2.3. Immune Evasion
M. pneumoniae has multiple strategies to escape host immune responses in order to ensure survival of the pathogen. Its survival includes immune evasion which may play an important role in pathogenesis. Inadequate immune responses against the invading pathogen results in uncontrolled proliferation and host tissue damage[88].
2.3.1. Molecular Mimicry
The term molecular mimicry can be described simply as “pathogens sharing a structural relationship with the host are tolerated as self, just like constituents of the host” [2][88][89]. The immune response targets the pathogen-peptide mimicking the host’s self-antigen, leading to the activation of naive, autoreactive T-cells specific to the corresponding self-antigen[89]. M. pneumoniae antigen mimics host cell components, thus the host immune response induced by the pathogen causes auto-immune responses and injuries to multiple organs[2][90].
The C-terminal region of the P1 and P30 proteins in M. pneumoniae show high levels of homology to troponin, cytoskeletal proteins, keratin and fibrinogen of the host[46][91]. Antibodies produced in response to M. pneumoniae infections will target various host tissues and form immune complexes, which aggravates the autoimmune response, leading to inflammatory injuries in the extrapulmonary tissues[13][46].
2.3.2. IbpM
Immunoglobin binding protein (IbpM) is a surface protein encoded by MPN400 that binds strongly to various immunoglobulins (IgM, IgG, and IgA) produced by the host[45]. Blötz et al. demonstrated that IbpM was required by M. pneumoniae to produce cytotoxic effects in host cells and is thus regarded as a virulent factor[45].
2.3.3. Antigen Variation
It has been observed that the surface adhesins P1, P40, and P90 of Mycoplasma pneumoniae display sequence variation[92][93]. Sluijter et al. demonstrated that the RecA protein homolog encoded by MPN490 promoted gene exchange between homologous DNA sequences (RepMP) in M. pneumoniae[94]. The RepMP are repetitive sequences present within genes encoding surface proteins such as the adhesins. Homologous recombination between these RepMP sequences generates sequence changes within the adhesin genes, which results in variations of surface adhesins and facilitates evasion of host immune surveillance[94][95][96].
The role of post-translational modifications of M. pneumoniae-specific proteins (e.g., P1, P40, P90) is a relatively new aspect of bacterial epigenetics[34]. The posttranslational modification of cytoadherence proteins by the protein kinase PrkC is essential for the development and function of the M. pneumoniae terminal organelle[97]. P1 adhesin of M. pneumoniae M129 is subject to extensive post-translational processing forming 22 proteo-forms, which are specific molecular forms of a protein product arising from a specific gene. Each of the proteo-forms retain the ability to bind to host molecules or their structural mimics and are surface accessible[31]. There are many issues that require further study, such as whether the antigen variations caused by post-translational modifications can affect the pathogenicity of M. pneumoniae.
2.3.4. Intracellular Survival
M. pneumoniae can survive for a long time in the human lung carcinoma cell (A549)[98], but the pathways related to intracellular survival remain to be elucidated. Intracellular M. pneumoniae has mechanisms to protect the pathogen against phagocytosis and antibiotics. This may explain why M. pneumoniae infection can develop into chronic lung disease, such as refractory pneumonia caused by macrolide-resistant M. pneumoniae due to the lack of timely and effective antibiotic treatment.
2.3.5. Others
Moreover, M. pneumoniae has an antioxidant mechanism to protect against oxidative reactions such as reactive oxygen species (ROS) damage[46][99]; A nuclease encoded by MPN491 can degrade neutrophil extracellular traps (NETs), which helps the pathogen to escape from the immune attack of host cells[44].
In summary, the pathogenesis of M. pneumoniae involves mainly the following four factors: immune evasion, adhesion, inflammatory injury and cytotoxicity. Figure 2 shows these four key pathogenic mechanisms of M. pneumoniae infection.
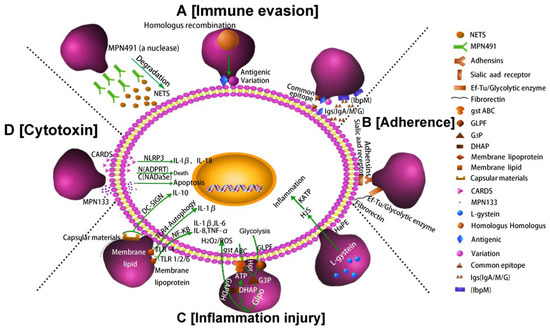
Figure 2. Pathogenic mechanisms of M. pneumoniae. (A) Nuclease and IbpM in M. pneumoniae enable immune evasion, and homologous DNA recombination leads to antigen variation; (B) M. pneumoniae adhesion causes cell damage. Additionally, the P1 adhesin protein binds to the sialic acid receptor on the host cell surface contributing to M. pneumoniae adherence and gliding. Furthermore, elongation factor Tu (EF-Tu) can bind strongly to a diverse range of host molecules (such as fibronectin), contributing to adhesion; (C) Inflammation-inducing factors (HapE enzyme, oxidase GlpO, membrane lipids, lipoproteins, and capsular materials) activate host cell inflammatory pathways; (D) M. pneumoniae secretes cytotoxic nuclease (catalytic protein encoded by MPN133) and CARDS toxin.
References
- Zhu, Y.G.; Tang, X.D.; Lu, Y.T.; Zhang, J.; Qu, J. Contemporary situation of community-acquired pneumonia in China: A systematic review. J. Transl. Int. Med. 2018, 6, 26–31.
- Meyer Sauteur, P.M.; Jacobs, B.C.; Spuesens, E.B.M.; Jacobs, E.; Nadal, D.; Vink, C.; Rossum, A.M.C. Antibody responses to Mycoplasma pneumoniae: Role in pathogenesis and diagnosis of encephalitis? PLoS Pathog. 2014, 10, e1003983.
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson1, E.J.; Courtney, D.M.; et al. Community-acquired pneumonia requiring Hospitalization among US adults. N. Engl. J. Med. 2015, 373, 415–427.
- Xiao, L.; Ptacek, T.; Osborne, J.D.; Crabb, D.M.; Simmons, W.L.; Lefkowitz, E.J.; Waites, K.B.; Atkinson, T.P.; Dybvig, K. Comparative genome analysis of Mycoplasma pneumoniae. BMC Genom. 2015, 16, 610.
- Xu, W.; Guo, L.; Dong, X.; Li, X.; Zhou, P.; Ni, Q.; Zhou, X.Y.; Wagner, B.L.; Li, L. Detection of viruses and Mycoplasma pneumoniae in hospitalized patients with severe acute respiratory infection in northern China, 2015–2016. Jpn. J. Infect. Dis. 2018, 71, 134–139.
- Kutty, P.K.; Jain, S.; Taylor, T.H.; Bramley, A.M.; Diaz, M.H.; Ampofo, K.; Arnold, S.R.; Williams, D.J.; Edwards, K.M.; McCullers, J.A.; et al. Mycoplasma pneumoniae among children hospitalized with community-acquired pneumonia. Clin. Infect. Dis. 2019, 68, 5–12.
- Waites, K.B.; Li, X.; Liu, Y.; Balish, M.F.; Atkinson, T.P. Mycoplasma pneumoniae from the respiratory tract and beyond. Clin. Microbiol. Rev. 2017, 30, 747–809.
- Søndergaard, M.J.; Friis, M.B.; Hansen, D.S.; Jørgensen, I.M. Clinical manifestations in infants and children with Mycoplasma pneumoniae infection. PLoS ONE 2018, 13, e0195288.
- Gao, L.W.; Yin, J.; Hu, Y.H.; Liu, X.Y.; Feng, X.L.; He, J.X.; Liu, J.; Guo, Y.; Xu, B.P.; Shen, K.L. The epidemiology of paediatric Mycoplasma pneumoniae pneumonia in North China: 2006 to 2016. Epidemiol. Infect. 2019, 147, e192.
- Kumar, S.; Roy, R.D.; Sethi, G.R.; Saigal, S.R. Mycoplasma pneumoniae infection and asthma in children. Trop. Dr. 2019, 49, 117–119.
- Kassisse, E.; García, H.; Prada, L.; Salazar, I.; Kassisse, J. Prevalence of Mycoplasma pneumoniae infection in pediatric patients with acute asthma exacerbation. Arch. Argent. Pediatr. 2018, 116, 179–185.
- Esposito, S.; Droghetti, R.; Bosis, S.; Claut, L.; Marchisio, P.; Principi, N. Cytokine secretion in children with acute Mycoplasma pneumoniae infection and wheeze. Pediatr. Pulmonol. 2002, 34, 122–127.
- Narita, M. Pathogenesis of extrapulmonary manifestations of Mycoplasma pneumoniae infection with special reference to pneumonia. J. Infect. Chemother. 2010, 16, 162–169.
- Narita, M. Classification of extrapulmonary manifestations due to Mycoplasma pneumoniae infection on the basis of possible pathogenesis. Front. Microbiol. 2016, 7, 23.
- Giavina-Bianchi, P.; Kalil, J. Mycoplasma pneumoniae infection induces asthma onset. J. Allergy Clin. Immunol. 2016, 137, 1024–1025.
- Wang, K.; Chalker, V.; Bermingham, A.; Harrison, T.; Mant, D.; Harnden, A. Mycoplasma pneumoniae and respiratory virus infections in children with persistent cough in England: A retrospective analysis. Pediatr. Infect. Dis. J. 2011, 30, 1047–1051.
- Takahashi, N.; Shinohara, T.; Oi, R.; Ota, M.; Toriumi, S.; Ogushi, F. Acute respiratory distress syndrome caused by Mycoplasma pneumoniae without elevated pulmonary vascular permeability: A case report. J. Thorac. Dis. 2016, 8, E319–E324.
- Meyer Sauteur, P.M.; Theiler, M.; Buettcher, M.; Seiler, M.; Weibel, L.; Berger, C. Frequency and clinical presentation of mucocutaneous disease due to Mycoplasma pneumoniae infection in children with community-acquired pneumonia. JAMA Dermatol. 2019, 156, 144–150.
- Meyer Sauteur, P.M.; Goetschel, P.; Lautenschlager, S. Mycoplasma pneumoniae and mucositis--part of the Stevens-Johnson syndrome spectrum. J. Dtsch. Dermatol. Ges. 2012, 10, 740–746.
- Harr, T.; French, L.E. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet. J. Rare. Dis. 2010, 5, 39.
- Prindaville, B.; Newell, B.D.; Nopper, A.J.; Horii, K.A. Mycoplasma pneumonia—Associated mucocutaneous disease in children: Dilemmas in classification. Pediatr. dermatol. 2014, 31, 670–675.
- Tabassum, I.; Chaudhry, R.; Chourasia, B.K.; Malhotra, P. Identification of an N-terminal 27 kDa fragment of Mycoplasma pneumoniae P116 protein as specific immunogen in M. pneumoniae infections. BMC Infect. Dis. 2010, 10, 350.
- Drasbek, M.; Nielsen, P.K.; Persson, K.; Birkelund, S.; Christiansen, G. Immune response to Mycoplasma pneumoniae P1 and P116 in patients with atypical pneumonia analyzed by ELISA. BMC Microbiol. 2004, 4, 7.
- Meyer Sauteur, P.M.; Unger, W.W.; Nadal, D.; Berger, C.; Vink, C.; van Rossum, A.M. Infection with and carriage of Mycoplasma pneumoniae in children. Front. Microbiol. 2016, 7, 329.
- De Groot, R.C.A.; Meyer Sauteur, P.M.; Unger, W.W.J.; van Rossum, A.M.C. Things that could be Mycoplasma pneumoniae. J. Infect. 2017, 74, S95–S100.
- Spuesens, E.B.M.; Meyer Sauteur, P.M.; Vink, C.; van Rossum, A.M.C. Mycoplasma pneumoniae infections-does treatment help? J. Infect. 2014, 69, S42–S46.
- Cao, B.; Qu, J.; Yin, Y.; Eldere, J.V. Overview of antimicrobial options for Mycoplasma pneumoniae pneumonia, focus on macrolide resistance. Clin. Respir. J. 2017, 11, 419–429.
- Khoury, T.; Sviri, S.; Rmeileh, A.A.; Nubani, A.; Abutbul, A.; Hoss, S.; van Heerden, P.V.; Bayya, A.E.; Hidalgo-Grass, C.; Moses, A.E.; et al. Increased rates of intensive care unit admission in patients with Mycoplasma pneumoniae: A retrospective study. Clin. Microbiol. Infect. 2016, 22, 711–714.
- Chen, Y.C.; Hsu, W.Y.; Chang, T.H. Macrolide-resistant Mycoplasma pneumoniae infections in pediatric community-acquired pneumonia. Emerg. Infect. Dis. 2020, 26, 1382–1391.
- Zhou, Y.; Zhang, Y.; Sheng, Y.; Zhang, L.; Shen, Z.; Chen, Z. More complications occur in macrolide-resistant than in macrolide-sensitive Mycoplasma pneumoniae pneumonia. Antimicrob. Agents Chemother. 2014, 58, 1034–1038.
- Widjaja, M.; Berry, I.J.; Jarocki, V.M.; Padula, M.P.; Dumke, R.; Djordjevic, S.P. Cell surface processing of the P1 adhesin of Mycoplasma pneumoniae identifies novel domains that bind host molecules. Sci. Rep. 2020, 10, 6384.
- Chang, H.Y.; Prince, O.A.; Sheppard, E.S.; Krause, D.C. Processing is required for a fully functional protein P30 in Mycoplasma pneumoniae gliding and cytadherence. J. Bacteriol. 2011, 193, 5841–5846.
- Chang, H.Y.; Jordan, J.L.; Kraus, D.C. Domain analysis of protein P30 in Mycoplasma pneumoniae cytadherence and gliding motility. J. Bacteriol. 2011, 193, 1726–1733.
- Widjaja, M.; Berry, I.J.; Pont, E.J.; Padula, M.P.; Djordjevic, S. P40 and P90 from Mpn142 are targets of multiple processing events on the surface of Mycoplasma pneumoniae. Proteomes 2015, 3, 512–537.
- Jordan, J.L.; Chang, H.Y.; Balish, M.F.; Holt, L.S.; Bose, S.R.; Hasselbring, B.M.; Waldo, R.H.; Krunkosky, T.M.; Krause, D.C. Protein P200 is dispensable for Mycoplasma pneumoniae hemadsorption but not gliding motility or colonization of differentiated bronchial epithelium. Infect. Immun. 2007, 75, 518–522.
- Chaudhry, R.; Varshney, A.K.; Malhotra, P. Adhesion proteins of Mycoplasma pneumoniae. Front. Biosci. 2007, 12, 690–699.
- Hasselbring, B.M.; Sheppard, E.S.; Krause, D.C. P65 truncation impacts P30 dynamics during Mycoplasma pneumoniae gliding. J. Bacteriol. 2012, 194, 3000–3007.
- Widjaja, M.; Harvey, K.L.; Hagemann, L.; Berry, I.J.; Jarocki, V.M.; Raymond, B.B.A.; Tacchi, J.L.; Gründe, A.; Steele1, J.R.; Padula, M.P.; et al. Elongation factor Tu is a multifunctional and processed moonlighting protein. Sci. Rep. 2017, 7, 11227.
- Balasubramanian, S.; Kannan, T.R.; Baseman, J.B. The surface-exposed carboxyl region of Mycoplasma pneumoniae elongation factor Tu interacts with fibronectin. Infect. Immun. 2008, 76, 3116–3123.
- Yu, Y.; Wang, H.; Wang, J.; Feng, Z.; Wu, M.; Liu, B.; Xin, J.; Xiong, Q.; Liu, M.; Shao, G. Elongation factor thermo unstable (EF-Tu) moonlights as an adhesin on the surface of Mycoplasma hyopneumoniae by binding to fibronectin. Front. Microbiol. 2018, 9, 974.
- Thomas, C.; Jacobs, E.; Dumke, R. Characterization of pyruvate dehydrogenase subunit B and enolase as plasminogen-binding proteins in Mycoplasma pneumoniae. Microbiology 2013, 159, 352–365.
- Dumke, R.; Hausner, M.; Jacobs, E. Role of Mycoplasma pneumoniae glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in mediating interactions with the human extracellular matrix. Microbiology 2011, 157, 2328–2338.
- Cloward, J.M.; Krause, D.C. Loss of co-chaperone TopJ impacts adhesin P1 presentation and terminal organelle maturation in Mycoplasma pneumoniae. Mol. Microbiol. J. 2011, 81, 528–539.
- Yamamoto, T.; Kida, Y.; Sakamoto, Y.; Kuwano, K. Mpn491, a secreted nuclease of Mycoplasma pneumoniae, plays a critical role in evading killing by neutrophil extracellular traps. Cell Microbiol. 2017, 19, e12666.
- Blötz, C.; Singh, N.; Dumke, R.; Stülke, J. Characterization of an immunoglobulin binding protein (IbpM) from Mycoplasma pneumoniae. Front. Microbiol. 2020, 11, 685.
- He, J.; Liu, M.; Ye, Z.; Tan, T.; Liu, X.; You, X.; Zeng, Y.H.; Wu, Y.M. Insights into the pathogenesis of Mycoplasma pneumoniae. Mol. Med. Rep. 2016, 14, 4030–4036.
- Großhennig, S.; Ischebeck, T.; Gibhardt, J.; Busse, J.; Feussner, I.; Stülke, J. Hydrogen sulfide is a novel potential virulence factor of Mycoplasma pneumoniae: Characterization of the unusual cysteine desulfurase/desulfhydrase HapE. Mol. Microbiol. 2016, 100, 42–54.
- Li, S.; Xue, G.; Zhao, H.; Feng, Y.; Yan, C.; Cui, J.; Sun, H.M. The Mycoplasma pneumoniae HapE alters the cytokine profile and growth of human bronchial epithelial cells. Biosci. Rep. 2019, 39, BSR20182201.
- Maenpuen, S.; Watthaisong, P.; Supon, P.; Sucharitakul, J.; Parsonage, D.; Karplus, P.A.; Claiborne, A.; Chaiyen, P. Kinetic mechanism of L-α-glycerophosphate oxidase from Mycoplasma pneumoniae. FEBS J. 2015, 282, 3043–3059.
- Shimizu, T.; Kimura, Y.; Kida, Y.; Kuwano, K.; Achibana, M.; Hashino, M.; Watarai, M. Cytadherence of Mycoplasma pneumoniae induces inflammatory responses through autophagy and Toll-like receptor 4. Infect. Immun. 2014, 82, 3076–3086.
- Choi, S.Y.; Lim, J.W.; Shimizu, T. Reactive oxygen species mediate Jak2/Stat3 activation and IL-8 expression in pulmonary epithelial cells stimulated with lipid-associated membrane proteins from Mycoplasma pneumoniae. Inflamm. Res. 2012, 61, 493–501.
- Wilson, M.H.; Collier, A.M. Ultrastructural study of Mycoplasma pneumoniae in organ culture. J. Bacteriol. 1976, 125, 332–339.
- Becker, A.; Kannan, T.R.; Taylor, A.B.; Pakhomova, O.N.; Zhang, Y.; Somarajan, S.R.; Galaleldeen, A.; Holloway, S.P.; Baseman, J.B.; Hart, P.J. Structure of CARDS toxin, a unique ADP-ribosylating and vacuolating cytotoxin from Mycoplasma pneumoniae. Proc. Natl. Acad. Sci. USA 2015, 112, 5165–5170.
- Kannan, T.R.; Krishnan, M.; Ramasamy, M.; Becker, M.; Pakhomova, O.M.; Hart, P.H.; Baseman, J.B. Functional mapping of community-acquired respiratory distress syndrome (CARDS) toxin of Mycoplasma pneumoniae defines regions with ADP-ribosyltransferase, vacuolating and receptor-binding activities. Mol. Microbiol. 2014, 93, 568–581.
- Somarajan, S.R.; Kannan, T.R.; Baseman, J.B. Mycoplasma pneumoniae Mpn133 is a cytotoxic nuclease with a glutamic acid-, lysine- and serine-rich region essential for binding and internalization but not enzymatic activity. Cell. Microbiol. 2010, 12, 1821–1831.
- Kawakita, Y.; Kinoshita, M.; Furukawa, Y.; Tulum, I.; Tahara, Y.O.; Katayama, E.; Namba, K.; Miyata, M. Structural study of MPN387, an essential protein for gliding motility of a human-pathogenic bacterium, Mycoplasma pneumoniae. J. Bacteriol. 2016, 198, 2352–2359.
- Hasselbring, B.M.; Krause, D.C. Proteins P24 and P41 function in the regulation of terminal-organelle development and gliding motility in Mycoplasma pneumoniae. J. Bacteriol. 2007, 189, 7442–7449.
- Nakane, D.; Kenri, T.; Matsuo, L.; Miyata, M. Systematic structural analyses of attachment organelle in Mycoplasma pneumoniae. PLoS Pathog. 2015, 11, e1005299.
- Miyata, M.; Hamaguchi, T. Integrated information and prospects for gliding mechanism of the pathogenic bacterium Mycoplasma pneumoniae. Front. Microbiol. 2016, 7, 960–976.
- Miyata, M.; Ogaki, H. Cytoskeleton of mollicutes. J. Mol. Microbiol. Biotechnol. 2006, 11, 256–264.
- Williams, C.R.; Chen, L.; Driver, A.D.; Arnold, E.A.; Sheppard, E.S.; Locklin, J.; Krause, D.C. Sialylated receptor setting influences Mycoplasma pneumoniae attachment and gliding motility. Mol. Microbiol. 2018, 109, 735–744.
- Krause, D.C.; Balish, M.F. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol. Lett. 2001, 198, 1–7.
- Chaudhry, R.; Ghosh, A.; Chandolia, A. Pathogenesis of Mycoplasma pneumoniae: An update. Indian J. Med. Microbiol. 2016, 34, 7–16.
- Balish, M.F. Mycoplasma pneumoniae, an underutilized model for bacterial cell biology. J. Bacteriol. 2014, 196, 3675–3682.
- Kang, M.; Hashimoto, A.; Gade, A.; Akbarali, H.I. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver. Physiol. 2015, 308, G532–G539.
- Großhennig, S.; Schmidl, S.R.; Schmeisky, G.; Busse, J.; Stülke, J. Implication of glycerol and phospholipid transporters in Mycoplasma pneumoniae growth and virulence. Infect. Immun. 2013, 81, 896–904.
- Merzbacher, M.; Detsch, C.; Hillen, W.; Stülke, J. Mycoplasma pneumoniae HPr kinase/phosphorylase. Eur. J. Biochem. 2004, 271, 367–374.
- Halbedel, S.; Hames, C.; Stülke, J. Regulation of carbon metabolism in the Mollicutes and its relation to virulence. J. Mol. Microb. Biotech. 2007, 12, 147–154.
- Into, T.; Dohkan, J.; Inomata, M.; Nakashima, M.; Shibata, K.I.; Matsushita, K. Synthesis and characterization of a dipalmitoylated lipopeptide derived from paralogous lipoproteins of Mycoplasma pneumoniae. Infect. Immun. 2007, 75, 2253–2259.
- Hallamaa, K.M.; Browning, G.F.; Tang, S.L. Lipoprotein multigene families in Mycoplasma pneumoniae. J. Bacteriol. 2006, 188, 5393–5399.
- Hallamaa, K.M.; Tang, S.L.; Ficorilli, N.; Browning, G.F. Differential expression of lipoprotein genes in Mycoplasma pneumoniae after contact with human lung epithelial cells, and under oxidative and acidic stress. BMC Microbiol. 2008, 8, 124.
- Into, T.; Kiura, K.; Yasuda, M.; Kataoka, H.; Inoue, N.; Hasebe, A.; Takeda, K.; Akira, S.; Shibata, K. Stimulation of human Toll-like receptor (TLR) 2 and TLR6 with membrane lipoproteins of Mycoplasma fermentans induces apoptotic cell death after NF-kappa B activation. Cell Microbiol. 2004, 6, 187–199.
- Hu, Q.; Zhao, Y.; Wang, Z.; Hou, Y.; Bi, D.; Sun, J.; Peng, X. Chicken gga-miR-19a targets ZMYND11 and plays an important role in host defense against Mycoplasma gallisepticum (HS strain) infection. Front. Cell. Infect. Microbiol. 2016, 6, 102.
- Kannan, T.R.; Provenzano, D.; Wright, J.R.; Baseman, J.B. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect. Immun. 2005, 73, 2828–2834.
- Kannan, T.R.; Baseman, J.B. ADP-ribosylating and vacuolating cytotoxin of Mycoplasma pneumoniae represents unique virulence determinant among bacterial pathogens. Proc. Natl. Acad. Sci. USA 2006, 103, 6724–6729.
- Krishnan, M.; Kannan, T.R.; Baseman, J.B. Mycoplasma pneumoniae CARDS toxin is internalized via clathrin-mediated endocytosis. PLoS ONE 2013, 8, e62706.
- Saber, S.; Ghanim, A.M.H.; El-Ahwany, E.; El-Kader, E.M.A. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343.
- Bose, S.; Segovia, J.A.; Somarajan, S.R.; Chang, T.H.; Kannan, T.R.; Baseman, J.B. ADP-ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. mBio 2014, 5, e02186-14.
- Yin, H.; Guo, Q.; Li, X.; Tang, T.; Li, C.; Wang, H.; Sun, Y.; Feng, Q.; Ma, C.; Gao, C. Curcumin suppresses IL-1β secretion an prevents inflammation through inhibition of the NLRP3 Inflammasome. J. Immunol. 2018, 200, 2835–2846.
- Hardy, R.D.; Coalson, J.J.; Peters, J.; Chaparro, A.; Techasaensiri, C.; Cantwell, A.M.; Kannan, T.R.; Baseman, J.B.; Dube, P.H. Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS ONE 2009, 4, e7562.
- Medina, J.L.; Coalson, J.J.; Brooks, E.G.; Winter, V.T.; Chaparro, A.; Principe, M.F.; Kannan, T.R.; Baseman, J.B.; Dube, P.H. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am. J. Respir. Cell. Mol. Biol. 2012, 46, 815–822.
- Maselli, D.J.; Medina, J.L.; Brooks, E.G.; Coalson, J.J.; Kannan, T.R.; Winter, V.T.; Principe, M.; Cagle, M.P.; Baseman, J.B.; Dube, P.H.; et al. The immunopathologic effects of Mycoplasma pneumoniae and community-acquired respiratory distress syndrome toxin: A primate model. Am. J. Respir. Cell. Mol. Biol. 2018, 58, 253–260.
- Shimizu, T.; Kida, Y.; Kuwano, K. Cytoadherence-dependent induction of inflammatory responses by Mycoplasma pneumoniae. Immunology 2011, 133, 51–61.
- Fang, X.; Liu, X.; Meng, C.; Fu, Y.; Wang, X.; Li, B.; Tu, F.; Zhao, F.; Ren, S. Breed-linked polymorphisms of porcine toll-like receptor 2 (TLR2) and TLR4 and the primary investigation on their relationship with prevention against Mycoplasma pneumoniae and bacterial LPS challenge. Immunogenetics 2013, 65, 829–834.
- Allen, P.Z.; Prescott, B. Immunochemical studies on a Mycoplasma pneumoniae polysaccharide fraction: Cross-reactions with type 23 and 32 antipneumococcal rabbit sera. Infect. Immun. 1978, 20, 421–429.
- Brunner, H. Protective efficacy of Mycoplasma pneumoniae polysaccharides. Isr. J. Med. Sci. 1981, 17, 678–681.
- Simmons, W.L.; Daubenspeck, J.M.; Osborne, J.D.; Balish, M.F.; Waites, K.B.; Dybvig, K. Type 1 and type 2 strains of Mycoplasma pneumoniae form different biofilms. Microbiology 2013, 159, 737–747.
- Root-Bernstein, R.; Fairweather, D. Complexities in the relationship between infection and autoimmunity. Current allergy and asthma reports. Curr. Allergy. Asthma. Rep. 2014, 14, 407.
- Sfriso, P.; Ghirardello, A.; Botsios, C.; Tonon, M.; Zen, M.; Bassi, N.; Bassetto, F.; Doria, A. Infections and autoimmunity: The multifaceted relationship. J. Leukoc. Biol. 2010, 87, 385–395.
- Saraya, T.; Kurai, D.; Nakagaki, K.; Sasaki, Y.; Niwa, S.; Tsukagoshi, H.; Nunokawa, H.; Ohkuma, K.; Tsujimoto, N.; Hirao, S.; et al. Novel aspects on the pathogenesis of Mycoplasma pneumoniae pneumonia and therapeutic implications. Front. Microbiol. 2014, 5, 410.
- Dallo, S.F.; Chavoya, A.; Baseman, J.B. Characterization of the gene for a 30-kilodalton adhesion-related protein of Mycoplasma pneumoniae. Infect. Immun. 1990, 58, 4163–4165.
- Kenri, T.; Taniguchi, R.; Sasaki, Y.; Okazaki, N.; Narita, M.; Izumikawa, K.; Umetsu, M.; Sasaki, T. Identification of a new variable sequence in the P1 cytadhesin gene of Mycoplasma pneumoniae: Evidence for the generation of antigenic variation by DNA recombination between repetitive sequences. Infect. Immun. 1999, 67, 4557–4562.
- Spuesens, E.B.M.; Hartwig, N.G.; van Rossum, A.M.C.; Vink, C. Sequence variation within the P1 gene of Mycoplasma pneumoniae. J. Clin. Microbiol. 2011, 49, 3723–3724.
- Sluijter, M.; Spuesens, E.B.M.; Hartwig, N.G.; van Rossum, A.M.C.; Vink, C. The Mycoplasma pneumoniae MPN490 and Mycoplasma genitalium MG339 genes encode recA homologs that promote homologous DNA strand exchange. Infect. Immun. 2009, 77, 4905–4911.
- Zhao, F.; Cao, B.; Li, J.; Song, S.; Tao, X.; Yi, Y.; He, L.; Zhang, J. Sequence analysis of the P1 adhesin gene of Mycoplasma pneumoniae in clinical isolates collected in Beijing in 2008 to 2009. J. Clin. Microbiol. 2011, 49, 3000–3003.
- Nakane, D.; Adan-Kubo, J.; Kenri, T.; Miyata, M. Isolation and characterization of P1 adhesin, a leg protein of the gliding bacterium Mycoplasma pneumoniae. J. Bacteriol. 2011, 193, 715–722.
- Schmidl, S.R.; Gronau, K.; Hames, C.; Busse, J.; Becher, D.; Hecker, M.; Stülke, J. The stability of cytadherence proteins in Mycoplasma pneumoniae requires activity of the protein kinase PrkC. Infect. Immun. 2010, 78, 184–192.
- Yavlovich, A.; Tarshis, M.; Rottem, S. Internalization and intracellular survival of Mycoplasma pneumoniae by non-phagocytic cells. FEMS Microbiol. Lett. 2004, 233, 241–246.
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Bio. Med. 2014, 66, 75–87.

