 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Konrad Sandhoff | + 4112 word(s) | 4112 | 2020-04-15 09:37:07 | | | |
| 2 | Rita Xu | -1379 word(s) | 2733 | 2020-04-26 10:21:11 | | | | |
| 3 | Rita Xu | -57 word(s) | 2676 | 2020-10-29 09:01:17 | | |
Video Upload Options
Gangliosides are sialic acid containing complex glycolipids, anchored and enriched in the outer leaflet of neuronal plasma membranes, with their glycan chains facing the extracellular space. Undegradeable gangliosides and related glycosphingolipids and oligosaccharides accumulate progressively in fatal lysosomal storage diseases, originally described as infantile amaurotic idiocy. Their lysosomal storage is caused by specific monogenic defects of catabolic hydrolyses or ancillary lipid-binding and -transfer proteins, essential for specific steps in their lysosomal catabolism.
However, small gangliosides can also accumulate as secondary material in other lysosomal storage diseases without a known defect in their catabolic pathway. Primary storage material of such diseases, sphingomyelin, lysosphingolipids, cholesterol and chondroitin sulfate are efficient inhibitors of specific steps of ganglioside catabolic pathway. They can attenuate ganglioside turnover, assisted by lipid binding proteins, the GM2 activator protein (GM2AP) and saposin B
1. Primary Storage Compounds in Gangliosidoses and Historical Aspects
The clinical picture of amaurotic idiocy, a fatal inherited form of progressive mental deficiency associated with early blindness, was known for a long time [1][2]. Pathological, histochemical, and chemical investigations eventually identified the accumulation of novel glycolipids in the brain tissue of infantile patients, named “Gangliosides” by Ernst Klenk [3]. Chemical, metabolic, enzymatic, and genetic investigations clarified the molecular, genetic, and cellular basis of these lysosomal storage diseases (LSDs). Currently, early therapeutic approaches are under way for these inherited neurodegenerative diseases, especially a gene replacement approach for infantile patients. Two types of ganglioside (GG) storage disorders are known (Table 1). GM1 gangliosidosis, caused by an inherited deficiency of the GM1 degrading lysosomal hydrolase acid β-galactosidase (EC 3.2.1.23), and several forms of GM2 gangliosidosis that result from defects in the GG GM2 catabolizing β-hexosaminidases and the hexosaminidases A (Hex A) associated GM2 activator protein (GM2AP), a lipid binding and transfer protein [4][5].
Table 1. Gangliosidoses.
| Disease |
Affected Protein
|
Affected Gene | Storage Compound | References |
|---|---|---|---|---|
| GM2 Gangliosidoses | ||||
| Tay–Sachs disease (B variant) | Hex A1, Hex S2 | HEXA | GM2, SM2a, lyso-GM2, GA2 | [6] |
| B1 variant | Hex A1 | HEXA | GM2 | [6] |
| Sandhoff disease | Hex A1, Hex B3 | HEXB | GM2, globoside, oligosaccharides, lyso-GM2 | [6][7][8] |
| GM2AP deficiency (AB variant) | GM2AP | GM2A | GM2 | [6] |
| GM1 Gangliosidosis | acid β-galactosidase | GLB1 | GM1, GA1, GM2, GM3, GA1a, lyso GM1 GlcCer Laccer, oligosaccharides, keratan sulfate | [9][10] |
1 Hex A αβ-subunit, 2 Hex S αα-subunit, and 3 Hex B ββ-subunit.
In 1881, Warren Tay [11] described an infantile patient with the clinical diagnosis of amaurotic idiocy, suffering from blindness and loss of its cognitive capabilities, now called Tay–Sachs disease (TSD). Its major neuronal storage compound, the GM2 (Table 1), is a degradation product of GG GM1 [12][13], the structure of which was elucidated in 1963 [14]. The elucidation of the enzyme-catalyzed GM2 hydrolysis was complex and tedious [15]. A Hex A-deficiency observed in the postmortem brain tissue of a single TSD patient in 1967 could not have been published. It was questioned by the analysis of another infantile “TSD patient” with GM2 storage in the brain that showed no enzyme deficiency, but an elevation of both, Hex A and Hex B activity levels in its postmortem brain tissue [16][17]. The latter patient was identified later as the first one to suffer from the defect of an essential lipid-binding cofactor of GG GM2 hydrolysis, the GM2AP, and not from an enzyme deficiency [18], see below.
After receiving postmortem material of two additional patients, we confirmed the Hex A deficiency in TSD [16], which was also demonstrated by [19]. In vitro experiments proved the basic assumption, that Hex A indeed splits GM2 slowly in the presence of an appropriate anionic detergent to release its terminal sugar to form the minor GG GM3 [20].
Two types of catabolic GG disorders are known (Table 1). GM1 gangliosidosis is caused by a deficiency of the GM1 degrading lysosomal hydrolases acid β-galactosidase (EC 3.2.1.23). GM2 gangliosidoses are a group of disorders that result from defects in digestion of GG GM2 and related glycolipids by β-hexosaminidases (EC 3.2.1.52) and the GM2AP [4][21]. Both gangliosidoses are progressive neurodegenerative diseases previously diagnosed as “amaurotic idiocy” [12][22].
1.1. GM2 Gangliosidoses
GM2 gangliosidoses comprise four inherited neurodegenerative disorders (TSD, B1 variant of GM2 gangliosidosis, Sandhoff disease (SD), and GM2AP deficiency (AB variant)). All of these disorders are based on defects in the degradation of GM2 and related glycolipids by the β-hexosaminidases (EC 3.2.1.52) and the GM2AP [17][22]. The β-hexosaminidases are a combination of two subunits (α or β). Hex A consisting of an α- and a β-subunit cleaves terminal β-glycosidically linked N-acetylglucosamine and N-acetylgalactosamine residues from negatively charged and uncharged glycoconjugates. Hex B contains two β-subunits (ββ) and Hex S two α-subunits (αα) (Table 1). Hex B splits uncharged substrates such as the GSLs GA2, globoside (Gb4Cer), and oligosaccharides with terminal N-acetylhexosamine residues. Hex S is involved in the degradation of glycosaminoglycans, sulfated glycolipid SM2a, and glycolipid GA2 and GM2 [23].
1.1.1. Tay–Sachs Disease (B Variant) and B1 Variant
TSD (B variant) is caused by α-chain mutations resulting in a deficiency of Hex A and Hex S activities, but keeps a normal Hex B activity [17]. The main neuronal storage compounds are the GG GM2 and its sialic acid free residue, the GSL GA2, minor accumulating metabolites are lyso-GM2 and SM2a [6].
Some patients generate a partially defective α-chain, which binds the β-subunit to generate a defective αβ-dimer (a defective Hex A), which is, however, still active against neutral substrates but has lost its catabolic activity against GM2 [24][25]. This variant is also called B1 variant of GM2-gangliosidosis. In contrast to TSD, Hex A of variant B1 patients is still active with neutral substrates like the glycosphingolipid GA2 and synthetic MUF (4-methylumbelliferone)-substrate, MUF-β-GlcNAc, but has lost its activity against anionic substrates like the main neuronal storage lipid, ganglioside GM2, and the soluble fluorogenic 4-methylumbelliferyl-6-sulfo-2-acetamido-2-deoxy-β-d-glycopyranoside (MUGS) substrate. Therefore, it differs from infantile TSD that lost Hex A activities against both, neutral and anionic substrates.
In contrast to the human clinical phenotype, mouse models for TSD differ severely in their phenotypes. The mouse model lacking Hex A and Hex S, shows no significant neurological phenotype. Mouse sialidase has some low activity, more than human enzyme, to accept GM2 as substrate, converting it slowly to GA2 [26], which is further degraded by the still active Hex B in the TSD mice.
In vitro studies have shown that sulfatide SM2a is also degraded by Hex A and Hex S in the presence of GM2AP [23]. Their functional deficiency triggers an accumulation of SM2a in TSD liver as well as in kidneys of the TSD mouse model [23].
1.1.2. Sandhoff Disease (SD) (0 Variant)
Another genetic variant of amaurotic idiocy with a clinical picture typical of TSD, but exposing additional visceral involvement and an additional lipid storage of globoside in the visceral organs was discovered in 1967 [7][8] and described as Variant 0 of GM2 gangliosidosis [17], now called “Sandhoff disease” (SD). It is characterized by an inherited deficiency of both major hexosaminidases, Hex A and Hex B, triggered by a genetic defect of their common β-subunit.
In SD, the functional loss of both, Hex A and Hex B, causes an increased storage of uncharged glycolipid GA2 in the brain besides GG GM2 and an additional accumulation of globoside (globotetraosylceramide) and oligosaccarides in visceral organs [17][22]. The clinical phenotype is similar to TSD, but with additional mucopolysacchariduria in visceral organs and secretion of oligosaccharides into the urine.
In accordance with infantile patients, the SD mouse, lacking Hex A and Hex B, shows a severe neurological phenotype [27][28].
1.1.3. GM2 Activator Protein Deficiency (AB Variant)
An ultra-rare neurodegenerative genetic disease with GM2 as the main neuronal storage compound is caused by an inherited deficiency of an essential lipid binding and transfer glycoprotein, the GM2AP, having, however, about normal levels of functional Hex A, Hex B, and Hex S activities [18]. The concept, that both are needed, a soluble hydrolase and a corresponding lipid binding or sphingolipid activator protein (SAP), was found to be correct for the physiological degradation of many sphingolipids (Figure 1) [4].
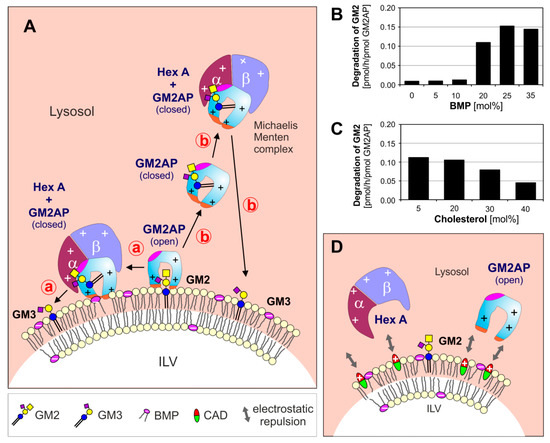
Figure 1. (A) Model for enzymatic digestion of membrane bound GM2 by Hex A, assisted by GM2AP at the surface of ILV at low, lysosomal pH-values. The open and empty GM2AP conformation binds to the membrane, e.g., by affinity to its lipid ligand and charge dependent interaction of the cationic protein (+, positively charged) with negatively charged membrane lipids (−, negatively charged) like BMP. Thereafter, the activator can interact with the ceramide portion of the GM2-ligand, which can move inside the hydrophobic cavity of the GM2AP, exposing the glycan chain of the GM2 to the water-soluble Hex A for hydrolysis. At this point, the conformation of the lipid-loaded activator may change to the closed one, thus the complex becomes more water soluble and lift the GG GM2 out of the membrane.The complex (GM2-GM2AP) can either stay at the surface of the membrane (pathway ⓐ) or leave the membrane (pathway ⓑ). (B, C) The GM2 hydrolysis is affected by membrane lipids: (B) anionic lipids e.g., BMP stimulate, sphingomyelin and (C) cholesterol inhibits GM2 degradation [29]. (D) CADs reaching the lysosome behave like cationic amphiphilic lipids, insert into the membrane surface of the intralysosomal luminal vesicles (ILVs) and start to compensate their negative surface charge. This results in a decreasing electrostatic attraction between proteins and ILVs, and an increasing repulsion between positively charged lysosomal proteins and the CAD-containing ILV-membrane. BMP: bis(monoacylglycero)phosphate, CADs: cationic amphiphilic drugs, Chol: cholesterol, GM2AP: GM2 activator protein, Hex A: β-hexosaminidase A, ILV: intralysosomal luminal vesicles.
1.2. GM1 Gangliosidosis
GM1 gangliosidosis was discovered by identifying GG GM1 and GSL GA1 as the main storage compounds in the postmortem brain tissue of a patient with infantile amaurotic idiocy [12][13]. The proposed block, a functionally deficient GM1-β-galactosidase in the catabolic pathway of GM1 was proven later by O’Brien et al. [22][30]. As many other lysosomal hydrolases, the GM1-β-galactosidase is a promiscuous glycosidase, which cleaves many oligosaccharides. Its deficiency triggers the accumulation of many other glycan substrates in GM1 gangliosidosis [31].
GM1 gangliosidosis is a progressive neurodegenerative disease due to absence or defective function of lysosomal acid β-galactosidase (E.C. 3.2.1.23), resulting in a storage of GM1 and its sialic acid-free derivate GA1. β-Galactosidase is part of a lysosomal multienzyme complex, containing also a sialidase and cathepsin A, the so-called protective protein. Another disease caused by mutations in the β-galactosidase gene GBL1 is mucopolysaccharidosis type IVB called Morquio type B disease. These mutations are different from those of GM1 gangliosidosis, and lead to a changed substrate specificity of the enzyme, thereby resulting in major accumulation of galactose containing keratan sulfate and oligosaccharides [21][32].
In vitro studies have shown that two SAPs, GM2AP and saposin (Sap) B, redundantly stimulate the GM1 hydrolysis by β-galactosidase [33]. Therefore, neither a defect of GM2AP nor of Sap B causes a GM1 accumulation, since the one remaining efficiently facilitates the reaction.
1.3. Topology and Regulation of GG Catabolism
Due to their lipid and amphiphilic nature and their poor solubility in aqueous solutions, GGs and other amphiphilic sphingolipids are membrane components of eukaryotic cells. GGs are especially enriched in neuronal plasma-membranes of mammalian cells. GGs are degraded after endocytosis and internalization into intralysosomal luminal vesicles (ILVs). Functional defects of any catabolic step cause an accumulation of membrane-bound-GGs in the lysosome. Based on experimental evidence obtained in vitro and by in vivo studies using murine and human cell cultures, we assume that the surface of ILVs, carrying GGs and other complex lipids, is the main location of their catabolism in the lysosomal compartment [4][34][35].
At low pH-values, most lysosomal hydrolases are protonated and positively charged, whereas the surfaces of the ILV membranes are negatively charged, mainly due to their high content of the anionic lysolipid bis(monoacylglycero)phosphate (BMP) and the possible presence of other anionic phospholipids in the ILV membranes [36]. BMP is an intermediate of the phosphatidylglycerol catabolism and can enrich in ILV membranes up to 40–60 mol% of their phospholipid content [4][37][38], mainly due to its slow catabolism. It can generate a negative zeta potential on the surfaces of ILVs [36], which electrostatically attracts positively charged hydrolases and SAPs to the sphingolipid-substrate carrying ILV-membranes, speeding up their catabolic rates [4][39][40] (Figure 1).
For diagnosis of LSDs, most lysosomal hydrolases are usually assayed in vitro with the help of synthetic and soluble fluorogenic substrates [41][42]. These convenient assays are an easy way to detect the presence of a lysosomal hydrolase in patients’ samples and to determine its activity in vitro. The activity measured in vitro with soluble synthetic substrates, however, does not indicate, in any way, the level of the sphingolipid-substrate cleaving activity of the patient’s hydrolase in vivo [43], since the sphingolipid cleaving activity of a lysosomal hydrolase can be strongly regulated and modified in vivo by genetic and by post translational modifiers which do not affect its activity against soluble substrates in vitro.
As genetic modifiers we consider the SAPs, small lipid-binding, lipid-transfer and/or vesicle-fusion glycoproteins of the lysosomal compartment. These cofactors are essential for the glycosphingolipid and sphingolipid cleaving activity of lysosomal hydrolases to reach physiologically relevant levels. Their inherited deficiencies can cause fatal storage diseases [4][34][35][44] despite the presence of fully active hydrolases, detectable with soluble fluorogenic substrates in patient`s cultivated cells or blood samples.
As posttranslational modifiers of the lipid-cleaving activity of lysosomal hydrolases we consider the strong inhibitory action of chondroitin-sulfate (accumulating in the lysosomes of Hurler, Hunter, Sanfilippo and Sly disease [45]) on the GM2 catabolism, as well as the collective properties of the sphingolipid-substrate carrying vesicle-membranes (e.g., extent of negative surface charge of ILVs to attract and bind protonated and positively charged hydrolases and the lipid composition of the vesicular membranes, especially the presence of stimulatory (e.g., BMP, ceramide) or inhibitory lipids (cholesterol, sphingomyelin)). These genetic and posttranslational modifiers strongly regulate the lipid-cleaving activity of lysosomal hydrolases, but rarely affect their activity against soluble, synthetic substrates used in vitro to diagnose lysosomal lipid storage diseases, as recently detailed in reconstitution experiments for the regulation of GM2 cleavage by Hex A in comparison to the unaffected cleavage of the soluble substrate MUGS (4-methylumbelliferyl-6-sulfo-2-acetamido-2-deoxy-β-D-glycopyranoside) [43].
Organellar membranes of eukaryotic cells maintain an organelle specific protein and lipid composition [46]. Cellular plasma membranes are rich in stabilizing lipids like cholesterol (up to 40 mol% of their lipid content), and maintain in their outer leaflet high levels of sphingomyelin and complex glycosphingolipids. Both, cholesterol and sphingomyelin, were identified as major inhibitors of key steps of lysosomal sphingolipid catabolism [4][34]. Therefore, the conversion of inhibitory sphingomyelin into stimulatory ceramide by acid sphingomyelinase (ASM) along the endocytose pathway at the level of late endosomes, and the removal of inhibitory cholesterol from nascent ILVs by two sterol-binding and -transfer proteins, Niemann–Pick disease protein C type 1 (NPC1) and NPC2, are essential to allow a physiological GG GM2, sphingolipid and GSL turnover [29][35][47][48].
1.4. Outlook
Lysosomal GG and GSL accumulation had been also observed in LSDs without a genetic defect in the GG catabolism. While the secondary accumulation of GG GM2 in Niemann-Pick diseases and in some mucopolysaccaridoses [49][50],could be based on a strong inhibitory effect of the primary storage compounds in these diseases (SM,cholesterol and chondroitin sulfate [43][45]) on the GG GM2 catabolism [35], the molecular mechanism causing a secondary accumulation of GG GM2, small GGs and glycosphingolipids in many other LSDs remains to be analysed: In sphingolipidoses, prosaposin deficiency, mucolipidoses (MLs), glycoproteinoses, neuronal ceroid lipofuscinoses (NCLs) and hereditary spastic paraplegia (HSP). In these diseases, mainly the GGs, GM2 and GM3, are accumulated. They are minor compounds (1–2% of the total gangliosides) of the healthy human brain and their proportion is even smaller in mice. A secondary accumulation of GM2 and GM3 in many LSDs, however, is associated with neuropathology. Therefore, the molecular basis of their secondary accumulation in these diseases should be investigated.
Though many ganglioside storage diseases are known for a long time, there is still no cure available. Enzyme replacement therapy (ERT) has been established for some LSDs not involving the central nervous system. In recent years, gene-replacement therapy has been successfully studied in animal models of gangliosidoses and is now applied to patients. It carries the promise of a cure, but still needs a long way to really cure progressive neurodegenerative diseases like gangliosidoses.
References
- Sachs, B; On arrested cerebral development with speciel reference to its cortical pathology. J. Neur. Ment. Dis. 1887, 14, 541–553.
- Sachs, B; A family form of idiocy, generally fatal, associated with early blindness (amaurotic family idiocy). Nerv. Ment. Dis. 1896, 21, 475–479.
- E. Klenk; Niemann–Pick’sche Krankheit und Amaurotische Idiotie. Hoppe-Seyler´s Zeitschrift für physiologische Chemie 1939, 262, 128-143, 10.1515/bchm2.1939.262.3-5.128.
- Breiden, B.; Sandhoff, K; Lysosomal glycosphingolipid storage diseases. Annu. Rev. Biochem. 2019, 88, 461–485.
- Konrad Sandhoff; Klaus Harzer; Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. The Journal of Neuroscience 2013, 33, 10195-10208, 10.1523/JNEUROSCI.0822-13.2013.
- Gravel, R.; Kaback, M.M.; Proia, R.L.; Sandhoff, K.; Suzuki, K.; Suzuki, K. The GM2 gangliosidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3827–3876.
- Sandhoff, K.; Andreae, U.; Jatzkewitz, H; Deficient hexosaminidase activity in an exceptional case of Tay–Sachs disease with additional storage of kidney globoside in visceral organs. Pathol Eur. 1968, 3, 278–285.
- H. Pilz; D. Muller; K. Sandhoff; V. Ter Meulen; Tay-Sachssche Krankheit mit Hexosaminidase-Defekt. DMW - Deutsche Medizinische Wochenschrift 1968, 93, 1833-1839, 10.1055/s-0028-1110836.
- Suzuki, K.; Chen, G.C; GM1-gangliosidosis (generalized gangliosidosis). Morphology and chemical pathology. Pathol Eur. 1968, 3, 389–408.
- Siegel, D.A.; Walkley, S.U; Growth of ectopic dendrites on cortical pyramidal neurons in neuronal storage diseases correlates with abnormal accumulation of GM2 ganglioside. J. Neurochem. 1994, 62, 1852–1862.
- Tay, W; Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans. Ophthalmol. Soc. 1881, 1, 55–57.
- H Jatzkewitz; K Sandhoff; On a biochemically special form of infantile amaturotic idiocy. Biochimica et Biophysica Acta 1963, 70, 354–356.
- Sandhoff, K. Die Amaurotische Idiotie des Menschen als Störung im Glykosphingolipidstoffwechsel. Doctoral Thesis, University of Munich, Munich, Germany, 1965.
- Kuhn, R.; Wiegandt, H; Die Konstitution der Ganglio-N-tetraose und des Gangliosids GI. Chem. Ber. 1963, 96, 866–880.
- Sandhoff, K; The GM2-gangliosidoses and the elucidation of the beta-hexosaminidase system. Adv. Genet. 2001, 44, 351–354.
- K Sandhoff; Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 1969, 4, 351–354.
- K. Sandhoff; K. Harzer; W. Wässle; H. Jatzkewitz; ENZYME ALTERATIONS AND LIPID STORAGE IN THREE VARIANTS OF TAY-SACHS DISEASE. Journal of Neurochemistry 1971, 18, 2469-2489, 10.1111/j.1471-4159.1971.tb00204.x.
- E. Conzelmann; K. Sandhoff; AB variant of infantile GM2 gangliosidosis: deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2. Proceedings of the National Academy of Sciences 1978, 75, 3979-3983, 10.1073/pnas.75.8.3979.
- S. Okada; J. S. O'brien; Tay-Sachs Disease: Generalized Absence of a Beta-D-N-Acetylhexosaminidase Component. Science 1969, 165, 698-700, 10.1126/science.165.3894.698.
- Sandhoff, K. The hydrolysis of Tay–Sachs ganglioside (TSG) by human N-acetyl-beta-D-hexosaminidase A. FEBS Lett. 1970, 11, 342–344.
- Roger Sandhoff; Heike Schulze; Konrad Sandhoff; Ganglioside Metabolism in Health and Disease. Progress in Molecular Biology and Translational Science 2018, 156, 1-62, 10.1016/bs.pmbts.2018.01.002.
- K. Sandhoff; H. Jatzkewitz; G. Peters; Die infantile amaurotische Idiotie und verwandte Formen als Gangliosid-Speicherkrankheiten. Die Naturwissenschaften 1969, 56, 356-362, 10.1007/bf00596925.
- Hepbildikler, S.T.; Sandhoff, R.; Kölzer, M.; Proia, R.L.; Sandhoff, K. Physiological substrates for human lysosomal beta -hexosaminidase S. J. Biol. Chem. 2002, 277, 2562–2572.
- H.J. Kytzia; U. Hinrichs; I. Maire; K. Suzuki; K. Sandhoff; Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity.. The EMBO Journal 1983, 2, 1201-1205, 10.1002/j.1460-2075.1983.tb01567.x.
- H J Kytzia; K Sandhoff; Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. Journal of Biological Chemistry 1985, 260, 7568–7572.
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K; et al. Mouse models of Tay–Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 11, 170–176.
- Mylvaganam Jeyakumar; David Smith; Elena Eliott-Smith; Mario Cortina Borja; Gabriele Reinkensmeier; Terry D. Butters; Thorsten Lemm; Konrad Sandhoff; V.Hugh Perry; Raymond A. Dwek; Frances M Platt; An Inducible Mouse Model of Late Onset Tay–Sachs Disease. Neurobiology of Disease 2002, 10, 201-210, 10.1006/nbdi.2002.0511.
- Sango, K.; McDonald, M.P.; Crawley, J.N.; Mack, M.L.; Tifft, C.J.; Skop, E.; Starr, C.M.; Hoffmann, A.; Sandhoff, K.; Suzuki, K.; et al. Mice lacking both subunits of lysosomal [beta]-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat. Genet. 1996, 14, 348–352.
- Susi Anheuser; Bernadette Breiden; Günter Schwarzmann; Konrad Sandhoff; Membrane lipids regulate ganglioside GM2 catabolism and GM2 activator protein activity. Journal of Lipid Research 2015, 56, 1747-1761, 10.1194/jlr.M061036.
- O’Brien, J.S.; Stern, M.B.; Landing, B.H.; O’Brien, J.K.; Donnell, G.N. Generalized gangliosidosis: Another inborn error of ganglioside metabolism? Am. J. Dis. Child. 1965, 109, 338–346.
- Roger Lawrence; Jeremy L. Van Vleet; Linley Mangini; Adam Harris; Nathan Martin; Wyatt Clark; Sanjay Chandriani; Jonathan H. Lebowitz; Roberto Giugliani; Alessandra D'azzo; Gouri Yogalingam; Brett E. Crawford; Characterization of glycan substrates accumulating in GM1 Gangliosidosis.. Molecular Genetics and Metabolism Reports 2019, 21, 100524, 10.1016/j.ymgmr.2019.100524.
- Gundo Wilkening; Thomas Linke; Gunther Uhlhorn-Dierks; Konrad Sandhoff; Degradation of Membrane-bound Ganglioside GM1. Journal of Biological Chemistry 2000, 275, 35814-35819, 10.1074/jbc.m006568200.
- Taeko Miyagi; Kohta Takahashi; Koji Yamamoto; Kazuhiro Shiozaki; Kazunori Yamaguchi; Biological and Pathological Roles of Ganglioside Sialidases. Progress in Molecular Biology and Translational Science 2018, 156, 121-150, 10.1016/bs.pmbts.2017.12.005.
- Roger Sandhoff; Konrad Sandhoff; Emerging concepts of ganglioside metabolism. FEBS Letters 2018, 592, 3835–3864.
- Bernadette Breiden; Konrad Sandhoff; Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. International Journal of Molecular Sciences 2020, 21, 2566, 10.3390/ijms21072566.
- Vincent O. Oninla; Bernadette Breiden; Jonathan O. Babalola; Konrad Sandhoff; Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. Journal of Lipid Research 2014, 55, 2606-2619, 10.1194/jlr.m054528.
- Toshihide Kobayashi; Marie-Hélène Beuchat; Margaret Lindsay; Sonia Frias; Richard D. Palmiter; Hitoshi Sakuraba; Robert G. Parton; Jean Gruenberg; Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nature 1999, 1, 113-118, 10.1038/10084.
- Wiebke Möbius; E. Van Donselaar; Y. Ohno-Iwashita; Y. Shimada; H. F. G. Heijnen; J. W. Slot; H. J. Geuze; Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 2003, 4, 222-231, 10.1034/j.1600-0854.2003.00072.x.
- Graf, C.G.F.; Schulz, C.; Schmälzlein, M.; Heinlein, C.; Mönnich, M.; Perkams, L.; Püttner, M.; Boos, I.; Hessefort, M.; Lombana Sanchez, J.N; et al. Synthetic glycoforms reveal carbohydrate-dependent bioactivity of human saposin D. Angew Chem. Int. Ed. Engl. 2017, 56, 5252–5257.
- Melanie Kölzer; Norbert Werth; Konrad Sandhoff; Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Letters 2004, 559, 96-98, 10.1016/s0014-5793(04)00033-x.
- Chunli Yu; Qin Sun; Hui Zhou; Enzymatic Screening and Diagnosis of Lysosomal Storage Diseases. North American Journal of Medicine and Science 2013, 6, 186–193, 10.7156/najms.2014.0604186.
- Galjaard, H. Genetic metabolic diseases: Early diagnosis and prenatal analysis; Elsevier-Noth Holland Biomedical Press: Amsterdam, The Netherlands, 1980.
- Susi Anheuser; Bernadette Breiden; Konrad Sandhoff; Membrane lipids and their degradation compounds control GM2 catabolism at intralysosomal luminal vesicles.. Journal of Lipid Research 2019, 60, 1099-1111, 10.1194/jlr.M092551.
- Thomas Kolter; Konrad Sandhoff; Lysosomal degradation of membrane lipids. FEBS Letters 2010, 584, 1700-1712, 10.1016/j.febslet.2009.10.021.
- Susi Anheuser; Bernadette Breiden; Konrad Sandhoff; Ganglioside GM2 catabolism is inhibited by storage compounds of mucopolysaccharidoses and by cationic amphiphilic drugs. Molecular Genetics and Metabolism 2019, 128, 75-83, 10.1016/j.ymgme.2019.04.007.
- Gerrit Van Meer; Dennis R. Voelker; Gerald W. Feigenson; Membrane lipids: where they are and how they behave. Nature Reviews Molecular Cell Biology 2008, 9, 112-124, 10.1038/nrm2330.
- Misbaudeen Abdul-Hammed; Bernadette Breiden; Matthew Adebayo; Jonathan O. Babalola; Günter Schwarzmann; Konrad Sandhoff; Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. Journal of Lipid Research 2010, 51, 1747-1760, 10.1194/jlr.M003822.
- Michael L. Wang; Massoud Motamed; Rodney Infante; Lina Abi-Mosleh; Hyock Joo Kwon; Michael S. Brown; Joseph L. Goldstein; Identification of Surface Residues on Niemann-Pick C2 Essential for Hydrophobic Handoff of Cholesterol to NPC1 in Lysosomes. Cell Metabolism 2010, 12, 166-173, 10.1016/j.cmet.2010.05.016.
- Jatzkewitz, H.; Pilz, H.; Sandhoff, K; Quantitative Bestimmungen von Gangliosiden und ihren Neuraminsäurefreien Derivaten bei infantilen, juvenilen und adulten Formen der amaurotischen Idiotie und einer spätinfantilen biochemischen Sonderform. Journal of Neurochemistry 1965, 12, 135-144, 10.1111/j.1471-4159.1965.tb06749.x.
- Walkley, S.U.; Vanier, M.T; Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta. 2009, 1793, 726–736.

