 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Grazyna Niewiadomska | + 4007 word(s) | 4007 | 2021-01-12 04:24:30 | | | |
| 2 | Vivi Li | + 2 word(s) | 4009 | 2021-01-21 04:40:30 | | |
Video Upload Options
Although the mechanisms of toxic activity of tau are not fully recognized, it is supposed that the tau toxicity is related rather not to insoluble tau aggregates but to its intermediate forms. It seems that neurofibrillar tangles (NFTs) themselves, despite being composed of toxic tau, are probably neither necessary nor sufficient for tau-induced neuronal dysfunction and toxicity. Tau oligomers (TauOs) formed during the early stages of tau aggregation are the pathological forms that play a key role in eliciting the loss of neurons and behavioral impairments in several neurodegenerative disorders called tauopathies. They can be found in tauopathic diseases, the most common of which is Alzheimer’s disease (AD). Evidence of co-occurrence of b-amyloid, α-synuclein, and tau into their most toxic forms, i.e., oligomers, suggests that these species interact and influence each other’s aggregation in several tauopathies. The mechanism responsible for oligomeric tau neurotoxicity is a subject of intensive investigation.
1. Introduction
Neurodegenerative diseases share many common pathomechanisms, the most prevalent of which is the death of specific populations of neurons as a result of disturbed protein metabolism, which leads to the deposition of pathological products of their metabolism in the central nervous system. Many data indicate that protein conformation disorders are the mechanism leading to both direct neurotoxicity and insufficiently effective elimination processes of these proteins. As a result, the products of their metabolism are deposited both in the intercellular space and inside neurons. Among intracellular aggregates, the most common are filamentous tangles (NFT, neurofibrillar tangles) [1], which are pathomorphological deposits of the tau protein resulting from disturbance of post-translational processes of this protein in the nerve cell. NFTs were first identified and described in the pathology of Alzheimer’s disease [2], but they are also present in the pathology of many other dementia diseases, which are collectively known as tauopathies.
In its physiological state, tau is a protein with many functions that can have both positive and negative consequences for the cell [3]. This is due to its ability to form complexes with many proteins and other elements in the cell, and specifically, it may participate in many signaling pathways that determine cell functioning and survival [4][5]. Conversely, disturbances related to (i) alternative splicing of the tau gene, (ii) abnormal interactions between individual post-translational modifications, (iii) the formation of pathological tau aggregates, (iv) interactions with elements of the cytoskeleton, and (v) the coexistence of pathologies of other proteins forming amyloids lead to the appearance of pathological forms of the tau protein [6]. It was assumed that NFTs were the cause of neuronal toxicity, since they correlate very well with cognitive decline and neuronal loss [7]. However, in some animal models overexpressing tau, neurodegeneration has been demonstrated in the absence of NFT pathology [8]. The formation of insoluble NFTs was suggested rather to be a protective mechanism than a necessary precursor of the neuronal death in tauopathies [9]. Based on the obtained data, the researchers suggest that the presence of tau protein deposits in the form of NFT not only does not have a toxic effect on cells but is likely to repair damage caused by free radicals in the course of neurodegenerative diseases. Overexpression of the tau protein in neurons reduces the level of oxidative stress and prevents apoptosis, regardless of the tau hyperphosphorylation process taking place in the cell [10]. The obtained data indicate that phosphorylation of the tau protein may be a specific cell response to oxidative stress and is neuroprotective [11].
A growing body of experimental data has shown that tau in the nervous system can occur in many forms that perform physiological functions, but it can also display a pathological activity [12]. Forms of tau that are probably not toxic are monomers (≈60 kDa), straight filaments, paired helical filaments (PHF), neurofibrillary tangles (NFT), and ghost tangles [13]. In contrast, some types of tau dimers/trimers (120–180 kDa), small soluble oligomers (300–500 kDa), and granular tau oligomers (~1800 kDa) possibly display toxic activities [14][15][16][17].
In the pathology of Alzheimer’s disease (AD), a recently debated issue has become to explain the role of tau oligomerization in the disease pathology. Tau protein oligomers are characterized by a secondary β-sheet structure and contain 3 or 4 repeats of the microtubule binding domain, and after reaching a size greater than 20 nm, they begin to aggregate and, as a result, form fibrillar forms [18][19]. Tau oligomers are composed primarily of monomeric or dimeric subunits of the highly phosphorylated or pathologically truncated tau protein [20]. However, assembly of two distinct dimers and higher-order oligomers from full-length tau was also observed [21]. Granular tau oligomers consisting of approximately 40 tau protein molecules have also been identified in the brain tissue of AD patients. The results of the conducted research indicate that granular oligomers appear inside neurons even before the formation of PHF, because their presence is detected in the earliest stages [2] of Alzheimer’s disease [17][22].
Similarly, the role of tau monomers may be ambiguous, as their potential toxicity cannot be ruled out when they undergo post-translational modifications (PTMs) in the protein maturation process. Several studies demonstrated that AD-associated PTMs of tau impairs neuronal function, structure, and viability [23]. When tau composed mostly of monomers is injected into the hippocampus, it reduces the number of synapses, causes a loss of synaptic vesicles, and exerts detrimental effects on the morphology and connectivity of newborn granule neurons of dental gyrus, and these effects correlate with impaired behavior [24]. In addition, tau modifications suppress compensatory responses to mitochondrial stress and thus lead to numerous metabolic disturbances and reduced energy production [25]. It seems that the mechanism of toxicity of modified monomers is different from that of oligomers. The intracerebral injection of toxic monomers cleaves endogenous tau by activating calpain and consequently triggers apoptotic neuronal death, whereas oligomers induce auto-aggregation of the endogenous tau by conformational changes of the protein regardless of its phosphorylation [23]. In addition to oligomers and modified monomers, also, truncated tau containing the N-terminal domain exhibits toxic properties, causing disrupted Ca2+-dependent glutamate release, perturbation in K+-evoked calcium dynamics, deterioration in presynaptic terminals, neuritic degeneration, microtubule collapse, and reduction of mitochondrial density [26].
The high toxicity of TauOs may be related to their presence in the extracellular space and their propagation through nerve connections [27]. The presence of tau in the extracellular space is the result of the leakage of tau deposits from inside the degenerating neuron. Tau can then exist as single molecules as well as in the form of low complexed aggregates/oligomers [28]. A possible mechanism for the appearance of tau in the extracellular space is the secretion of tau aggregates in micro vesicles such as exosomes and ectosomes [29][30] and its release by exocytosis in a chaperone-dependent manner. There is also evidence that most of the extracellular tau is not associated with vesicle secretion [31]. Truncated tau or oligomeric tau may also propagate between cells by tunneling nanotubes [32][33]. Once tau appears in the extracellular space, it may undergo endocytosis by surrounding cells and influence their functioning [34][35]. The results of the experiment of Pampuscenko et al. [36] showed that the extracellular self-assembly dimers–tetramers of the 1N4R tau isoform (one extra exon in the N-terminal domain and four tubulin binding motifs), but not monomers, caused neuronal death in mixed neuronal/glial cell cultures. In contrast, monomeric and pre-aggregated tau peptide with 4R repeats but lacking the N-terminal fragment did not reduce the viability of the cells, suggesting that tau oligomers’ neurotoxicity might be dependent on the presence of the N-terminal fragment in the molecule.
It is possible that the toxic effect of the extracellular tau species is based on their interaction with M1 and M3 muscarinic membrane receptors [37][38]. As a consequence, there is an increase in intracellular calcium [Ca2+] and cell die due to excitotoxicity [27][39]. Several forms of tau were tested in these studies such as recombinant human tau (isoform with three tubulin binding motifs and two extra exons in the N-terminal domain), tau fragment 2N, which contains the amino-terminal half of the tau protein tau fragment 3RC, which contains the three tubulin-binding motifs and the carboxyl-terminal region, and tau fragments 4R, 3R, i.e., four or three tubulin-binding motifs. It was shown that mainly the tau fragment 3RC containing the C-terminal region and comprising residues 391 to 407 of the tau molecule is responsible for the excessive influx of calcium ions into the neuroblastoma cells. Recent reports suggest that the neurotoxic effect of extracellular tau interaction with muscarinic receptors is dependent on the activity of alkaline phosphatase [40].
The data reviewed in the present paper highlight the toxic effect of oligomeric forms of the tau protein on selected intraneuronal functions such as the stability of the genome and the function of the nucleus, energy production and mitochondrial function, cell signaling and synaptic plasticity, the microtubule assembly, neuronal cytoskeleton and axonal transport, and the effectiveness of the protein degradation system (Figure 1).
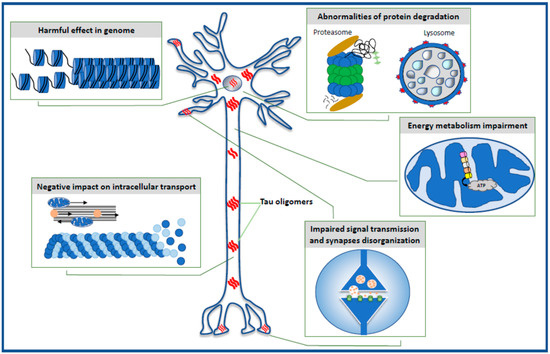
Figure 1. Schematic illustration of intraneuronal processes affected by toxic tau oligomers.
2. Harmful Effects in Genome
The presence of tau protein in the nucleus of neurons [41][42] and the capacity of tau protein to form protein–DNA complexes have been reported [41][43][44][45][46]. It has been proven that tau-interacting DNA regions are positioned at regular intervals in the structure of the chromosome. Tau protein plays a role also in the maintenance of genomic stability [47], protection of RNA and DNA from damage induced by oxidative stress [41][42][48], and in sustenance of the dense chromatin structure [49][50]. Dysfunctional nuclear tau may disrupt heterochromatin organization, leading to cell cycle re-entry, which is fatal to neurons, and dysregulate gene expression and RNA transcription, giving rise to altered protein synthesis. Post-translational modifications or conformational changes of the tau molecule can modulate its nuclear translocation and function [51]. An aberrant modification of tau in diseases such as AD could alter its function and enhance genome vulnerability and neurodegeneration [51].
Mansuroglu et al. [50] studied the physiological role of tau protein in maintaining the neuronal genome structure and organization, namely, the role of tau protein in the organization of pericentromeric heterochromatin (PCH) DNA regions. PCH contains very dense chromatin substructures rich in epigenetic factors, such as the trimethylated form of lysine 9 of histone H3 (H3K9me3) and the protein heterochromatin 1α (HP1α), which regulate genome expression and stability [52][53][54]. The studies were performed on primary cultures of neurons obtained from wild-type (WT) or tau-deficient (KOTau) mice. In WT mouse neurons, the tau protein was found within or in the immediate vicinity of PCH and interacting with major PCH satellite sequences [50]. Mansuroglu et al. [50] also showed that in the neurons of WT mice, H3K9me3 and HP1α were distributed in PCH in the form of regular clusters. This cluster distribution of H3K9me3 and HP1α was disrupted in the neurons of KOTau mice, although the total amount of H3K9me3 and HP1α remained unchanged compared to WT mice. Under heat stress condition, the disruption of PCH organization in the neurons of KOTau mice was associated with a high degree of DNA breaks accumulated mainly in PCH sequences [50]. At the same time, the loss of clustered distribution of H3K9me3 observed in KOTau neurons affected the localization of one of the histone proteins phosphorylated on serine 139 residue (γH2AX) and made KOTau neurons unable to repair PCH–DNA breakage induced by stress [50]. Interestingly, an induced overexpression of hTau in the nuclei of KOTau neurons restored the clustered distribution of H3K9me3 that was lost due to primary tau protein gene silencing. This observation confirmed the regulatory role of nuclear tau protein in the clustering distribution of H3K9me3 and HP1α in the structure of PCH.
The expression of γH2AX is an early cellular response to the induction of DNA double-strand breaks and is used as a highly specific and sensitive molecular marker to monitor the initiation of DNA damage [55]. Similar changes in γH2AX expression were reported in neurons from AD brains [56][57]. Mansuroglu et al. [50] suggested that changes in the structure of PCH, such as the disturbed localization pattern of H3K9me3, are partially responsible for the increased expression of heterochromatically silenced genes reported in the hippocampal neurons of AD patients [49].
H3K9me3 and γH2AX were detected not only in the nucleus but also as scattered small clusters in the cytoplasm of KOTau neurons [50]. This cytoplasmic distribution of H3K9me3 was also observed in neurons of AD patients and was co-localized with AT8 antibody staining, which detects phosphorylated forms of tau [58]. This may suggest that post-translational tau modifications affect the translocation of the protein between the nucleus and the cytoplasm and alter its physiological function.
Loss of tau nuclear physiological function may be caused by its oligomerization [59]. The physiological inhibitory effect of tau on gene expression has been demonstrated under heat shock conditions (HS) [60]. Tau appears to protect tau-interacting genes from deregulation that can occur during the HS-induced overexpression of these genes. The increased presence of pathological TauOs in the nuclear compartment of neurons was correlated with their inability to bind to DNA and their incapability to repress Tau-interacting genes [59]. The transcriptionally repressing role of Tau protein was augmented in neurons from the hippocampal CA1 region of THY-Tau22 mice, where a nuclear accumulation of pathological oligomerized forms of Tau protein was detected [59]. Tau oligomers were recognized by antibodies TOC1, AT100 for tau phosphorylated at residues 212 and 214, and Tau1au for unphosphorylated tau between residues 189 and 207. In this study, nuclear accumulation of pathological oligomerized forms of tau resulted in significant deregulation of the Dlg2 gene (coding Discs Large Scaffold Protein 2) expression in neurons from the CA1 region of THYTau22 mice. Dlg2 gene is connected with long non-coding (lnc)RNA and contains the highest number of tau-interacting sites, and it codes a key scaffolding protein for postsynaptic membranes (postsynaptic density protein 93-PSD-93) [61]. Deficiency of this gene product may cause disturbances in the clustering of membrane receptors, the permeability of ion channels, and associated synapse signaling.
Tau oligomers can interact with p53, which is a transcription factor involved in many processes such as apoptosis [62], DNA damage repair [63], and cell cycle control [63]. Farmer et al. [64] proved that the p53 protein and TauOs (recognized by the T22 antibody) interact in the neurons of AD patients and a mouse model associated with AD (Tg2576/Tau P301L). The evidences presented in the study by Farmer et al. [64] suggest that the interaction between p53 and TauOs may be due to protein misfolding. In the p53 molecule, the DNA-binding domains, the N-terminal domain and the C-terminal base domain, were in a state of intrinsic disorder [65][66] and therefore prone to misfolding. Tau is also intrinsically disordered [67] and therefore contained many aggregation-prone regions that could interact with p53. It is known that p53 is normally associated with microtubules [68]. Thus, the proximity of p53 and tau may increase the likelihood of p53 interaction with tau, also at an earlier stage of pathology, when TauOs are initially formed. The interaction of p53 and TauOs leading to the aggregation of both proteins inhibits nuclear import of the p53 and causes loss of its function of the transcription factor [64]. Lasagna-Reeves et al. [69] showed that aggregated p53 cannot bind to DNA, confirming that aggregation leads to a loss of p53 function. Moreover, the structure of the nuclear membrane and its pores can be affected by pathological forms of tau [70]. This may hinder p53 transport into the nucleus and cause its accumulation in the cytoplasm, which has been observed both in neurons of AD patients and tau overexpressing mice [64][71].
The nucleotoxic activity of TauOs may be related to the dysfunction of the Musashi family proteins (MSI1 and MSI2). MSIs belong to RNA-binding proteins involved in the transcription and post-transcriptional regulation of genes [72]. Montalbano et al. [73] observed oligomeric forms of MSI1 and MSI2 in the cytoplasm and in the nuclei of cortical neurons of AD, ALS (amyotrophic lateral sclerosis), and FTD (frontotemporal dementia) patients, where they were localized with oligomeric species of tau. Tau oligomers were identified with the TOMA2 antibody (anti-oligomeric tau monoclonal antibody). Montalbano et al. [73] also conducted studies on two iHEK (immortalized human epidermal keratinocytes) cell lines that express human wild-type tau or the form of tau containing the P301L mutation. In the P301L line, increased associations were observed between MSI2, toxic TauOs, Histone3, and Lamin (nuclear fibrous protein). This large complex can damage the chromatin structure and disarray its compartmentalization in the nucleus. In cells overexpressing the mutant form of tau, a significant decrease in the levels of the proteins LaminB1 and LaminA/C in the nuclear membrane was observed. This was accompanied by a general downregulation of nuclear membrane linkers such as emerin, LaminB receptor, companion lamellar protein 3 Man1 containing the polypeptide 2-emerin domain, and the nuclear envelope type II transmembrane protein Nesprin2. This resulted in instability of the nuclear membrane. Chromatin linkers such as heterochromatin 1 protein (HP1), plaque 2-associated polypeptide (LAP-2), and autointegration barrier factor (BAF) were also reduced.
Disruptions in the structure of the nuclear membrane and the pore system caused by pathological forms of tau were also described in the studies by Montalbano et al. [73]. Toxic tau influenced nucleocytoplasmic import, preventing substances from penetrating into the nucleus through the nuclear pore complex. It has been shown that TauOs can bind to phenylalanine/glycine-rich nucleoporins Nup98 and Nup62 and affect the permeability of the nuclear pore complex. The authors [73] also observed a decreased level of Nup50 and Nup153 nucleoporins in the nuclear fraction and at the same time their increased level in the cytoplasm of P301L tau iHEK cells. Thus, tau oligomers by interacting with nuclear transport receptors can seriously dysregulate intra-nuclear transport.
Pathogenic tau directly contributes to the depletion of cAMP response element binding protein (CREB), which is a major regulator of gene transcription critical to neuronal survival, neural plasticity, learning, and memory [74]. A significant decrease in the level of CREB and phospho-CREB was noted in post mortem human AD brain tissue [75] and in cultures of hippocampal neurons derived from tau transgenic mice [76]. Mahoney et al. [77] investigated whether pathogenic forms of tau influenced nuclear Ca2+ levels, which are critical for CREB-dependent gene expression. The observations were carried out in neurons derived from induced pluripotent stem cells (iPSCs) of patients with sporadic AD and in the Drosophila melanogaster tauopathy model expressing human tau bearing the R406W mutation [78]. TauR406W is an autosomal dominant mutation that causes frontotemporal lobe degeneration (FTLD) [79]. It was found that the neuronal expression of human transgenic tauR406W in the brain of adult Drosophila resulted in a reduction in the total and nuclear CREB protein level. It was observed that in the resting state, the level of nuclear Ca2+ decreased with physiological aging, and pathogenic tauR406W significantly increased the age-related loss of calcium ions from the nucleus. It has also been reported that blocking the nuclear Ca2+ signal enhances tauR406W-induced neuronal death, suggesting that Ca2+ nuclear depletion is a causal mediator of neurodegeneration in tauopathy [77].
3. Selective Effect of Tau Oligomers Treatment on Synaptic Integrity and Function
Since the discovery of tau mutation as a cause of FTLD, tau has been implicated in different synaptic dysfunctions, such as impaired LTP, reduced excitatory synaptic transmission, decreased level of PSD-95 and glutamate receptors, and increased levels of glutamate and excitotoxicity [80]. Usenovic et al. [81] noted that the model relevant for sporadic tauopathies, such as AD, in which the MAPT (microtubule-associated tau protein) mutations are absent would be one in which changes similar to those seen in AD would occur despite the absence of a mutation in the tau protein or lack of tau overexpression. They proposed such a model: the human forebrain neurons with active GABAergic and glutamatergic receptors, which were derived from induced pluripotent stem cells and treated with preparations of tau monomers and oligomers for 24 h. In this model, fourteen days after TauOs were applied, the number of synapses declined, basal GABA (gamma-aminobutyric acid) release moderately increased, and glutamate release remain unchanged. In addition to synaptic-related changes, they observed a host of other changes, which are similar to those characterizing AD. No such changes were observed when tau monomers were applied.
Effects of oligomeric tau treatment on synaptic integrity and function were also evident within a much shorter time frame. Lasagna-Reeves et al. [82] found in the neurons of the CA1 region of the hippocampus that the subcortical injection of TauOs reduced the levels of synaptophysin, which is a synaptic vesicle bound protein, and of septin-11, which is a protein involved in vesicle trafficking. On the other hand, the level of synapsin-1, the protein that regulates the reserve pool of synaptic vesicles, remained unchanged. These effects were observed about 30 h post-injection. The animals treated with TauOs did not distinguish the new object in the Object Recognition Task. This may mean that TauOs impair the ability to store newly acquired information, which is the kind of memory deficit occurring in early stage of AD. The injection of tau monomers and fibrillary forms neither affected the level of synaptic proteins nor caused memory deficit.
Fa et al. [83] showed that a brief exposure of mouse hippocampal slices to different recombinant human TauOs, but not tau monomers, reduced LTP as soon as 20 min after exposure to the oligomers. This finding was paralleled by the demonstration that two bilateral injections of TauOs performed shortly prior to the training into the dorsal mouse hippocampi impaired associative fear memory assessed 24 h later. The authors also demonstrated the worsening of spatial memory in a radial arm water maze after infusion of oligomeric tau.
Ondrejcak et al. [84] injected into brain ventricles soluble human recombinant tau aggregates and observed the potent inhibition of LTP at CA3 to CA1 synapses of the hippocampus, whereas no change in LTP followed the injection of tau monomers and tau fibrils. The gradual decline of LTP was observed during the 3 h period following tau injection. The effect of TauOs was prevented by the administration of anti-tau monoclonal antibody, Tau5 to the prion protein (PrP), which may suggest that the involvement of PrP in tau oligomers induced LTP reduction.
Hill et al. [85] delivered oligomeric full-length tau-441 (TauOs) in nanomolar concentration directly into mouse hippocampal CA1 pyramidal and neocortical layer V thick-tufted pyramidal cells. Within the first few minutes after intracellular TauOs delivery, the electrophysiological readouts remained unchanged, and some of them changed significantly after 40 min. The introduction of TauOs into hippocampal neurons increased the input resistance, membrane potential depolarization, and firing rate but decreased the amplitude, speed of rise, and speed of decay of action potentials. Examination of connected pairs of thick-tufted layer V neocortical pyramidal cells revealed that the delivery of TauOs into presynaptic cells impaired within a short time frame basal synaptic transmission, which was associated with short-term depression. The delivery of TauOs into postsynaptic cells impaired LTP, which may indicate that the presence of TauOs in postsynaptic cells interferes with synaptic plasticity. The delivery of tau monomers did not evoke any similar effects.
The observations presented above point collectively to TauOs being taken up by neurons from extracellular space, and once being inside the neurons, they change the functioning of synapses within a short time, even during tens of minutes. The relatively short time needed to reveal the effect of TauOs on various electrophysiological parameters, reported by Hill et al. [85], can be explained by the direct delivery of TauO into neurons, which shortened the time needed for TauO molecules to reach distal dendrites and axons, where they have been indeed clearly visible. These findings speak for the direct involvement of TauOs molecules in processes of synaptic transmission and plasticity. The common finding of reported data is the impairment of long-term potentiation (LTP). LTP is believed to be one of the cellular mechanisms that underlies learning and memory, and indeed, in the two studies [82][83], behavioral tests revealed TauOs were related to memory impairment.
The use of well-defined oligomeric forms of the tau protein in research shows that tau interacts with synaptic proteins. It plays a role in monitoring intracellular signaling pathways. The entering of pathological forms of tau into postsynaptic compartments and dendrites [80] causes a decrease in the number of synaptic vesicles [86] and reduction of synaptic proteins and dendritic spines, which in turn promotes synaptic loss [24], reduces neuronal signaling [87][88], and finally, induces memory impairment.
References
- Buée, L.; Bussičre, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130.
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76.
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704.
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426.
- Spires-Jones, T.L.; Stoothoff, W.H.; De Calingnon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159.
- Markesbery, W.R.; Schmitt, F.A.; Kryscio, R.J.; Davis, D.G.; Smith, C.D.; Wekstein, D.R. Neuropathologic substrate of mild cognitive impairment. Arch. Neurol. 2006, 63, 38–46.
- Feuillette, S.; Miguel, L.; Frébourg, T.; Campion, D.; Lecourtois, M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 2010, 113, 895–903.
- Bonda, D.J.; Castellani, R.J.; Zhu, X.; Nunomura, A.; Lee, H.G.; Perry, G.; Smith, M.A. A Novel Perspective on Tau in Alzheimer Disease. Curr. Alzheimer Res. 2011, 8, 639–642.
- Li, H.L.; Wang, H.H.; Liu, S.J.; Deng, Y.Q.; Zhang, Y.J.; Tian, Q.; Wang, X.C.; Chen, X.Q.; Yang, Y.; Zhang, J.Y.; et al. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 3591–3596.
- Castellani, R.J.; Nunomura, A.; Rolston, R.K.; Moreira, P.I.; Takeda, A.; Perry, G.; Smith, M.A. Sublethal RNA oxidation as a mechanism for neurodegenerative disease. Int. J. Mol. Sci. 2008, 9, 789–806.
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer’s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661.
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010, 49, 10039–10041.
- Fox, L.M.; William, C.M.; Adamowicz, D.H.; Pitstick, R.; Carlson, G.A.; Spires-Jones, T.L.; Hyman, B.T. Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J. Neuropathol. Exp. Neurol. 2011, 70, 588–595.
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013.
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201.
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular Tau oligomers as intermediates of Tau filaments. Biochemistry 2007, 46, 3856–3861.
- Sugino, E.; Nishiura, C.; Minoura, K.; In, Y.; Sumida, M.; Taniguchi, T.; Tomoo, K.; Ishida, T. Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochem. Biophys. Res. Commun. 2009, 385, 236–240.
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, L.; Rolkowa, G.; Kontsekova, E.; Nova, M. Truncated tau from sporadic Alzheimer’s diseases suffices to drive neurofibryllary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588.
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029.
- Maeda, S.; Takashima, A. Tau Oligomers. Adv. Exp. Med. Biol. 2019, 1184, 373–380.
- Manassero, G.; Guglielmotto, M.; Monteleone, D.; Vasciaveo, V.; Butenko, O.; Tamagno, E.; Arancio, O.; Tabaton, M. Dual Mechanism of Toxicity for Extracellular Injection of Tau Oligomers versus Monomers in Human Tau Mice. J. Alzheimers Dis. 2017, 59, 743–751.
- Bolós, M.; Pallas-Bazarra, N.; Terreros-Roncal, J.; Perea, J.R.; Jurado-Arjona, J.; Avila, J.; Llorens-Martin, M. Soluble tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl. Psychiatry 2017, 7, 1267.
- Guha, S.; Fischer, S.; Johnson, G.V.W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65.
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Maria Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778.
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Díaz-Hernández, J.I.; Miras-Portugal, M.T.; Avila, J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 2009, 19, 708–717.
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212.
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A New Mechanism for Non-Exosomal Secretion of Tau Protein. PLoS ONE 2014, 9, e100760.
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5.
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366.
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117.
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351.
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852.
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913.
- Pampuscenko, K.; Morkuniene, R.; Krasauskas, L.; Smirnovas, V.; Tomita, T.; Borutaite, V. Distinct Neurotoxic Effects of Extracellular Tau Species in Primary Neuronal-Glial Cultures. Mol. Neurobiol. 2020.
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell Neurosci. 2008, 37, 673–681.
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell Neurosci. 2019, 13, 403.
- Wysocka, A.; Palasz, E.; Steczkowska, M.; Niewiadomska, G. Dangerous Liaisons: Tau Interaction with Muscarinic Receptors. Curr. Alzheimer Res. 2020, 17, 224–237.
- Díaz-Hernández, M.; Gómez-Ramos, A.; Rubio, A.; Gómez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 2010, 285, 32539–32548.
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575.
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84.
- Sjöberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119, 2025–2034.
- Hua, Q.; He, R.Q.; Haque, N.; Qu, M.H.; Del Carmen Alonso, A.; Grundke-Iqbal, I.; Iqbal, K. Microtubule associated protein tau binds to double-stranded but not single-stranded DNA. Cell. Mol. Life Sci. 2003, 60, 413–421.
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE 2008, 3, e2600.
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533.
- Malmanche, N.; Dourlen, P.; Gistelinck, M.; Demiautte, F.; Link, N.; Dupont, C.; Vanden Broeck, L.; Werkmeister, E.; Amouyel, P.; Bongiovanni, A.; et al. Developmental expression of 4-Repeat-Tau induces neuronal aneuploidy in drosophila tauopathy models. Sci. Rep. 2017, 7.
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Sendid Chouala, M.; Sultan, A.; Marciniak, E.; Humez, S.; Binder, L.; Kayed, R.; et al. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis. 2015, 82, 540–551.
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366.
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 8.
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9.
- Brown, K.E.; Amoils, S.; Horn, J.M.; Buckle, V.J.; Higgs, D.R.; Merkenschlager, M.; Fisher, A.G. Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat. Cell Biol. 2001, 3, 602–606.
- Guenatri, M.; Bailly, D.; Maison, C.; Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 2004, 166, 493–505.
- Josse, T.; Mokrani-Benhelli, H.; Benferhat, R.; Shestakova, E.; Mansuroglu, Z.; Kakanakou, H.; Billecocq, A.; Bouloy, M.; Bonnefoy, E. Association of the interferon-β gene with pericentromeric heterochromatin is dynamically regulated during virus infection through a YY1-dependent mechanism. Nucleic Acids Res. 2012, 40, 4396–4411.
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686.
- Richards, E.J.; Elgin, S.C. Epigenetic codes for heterochromatin formation and silencing: Rounding up the usual suspects. Cell 2002, 108, 489–500.
- Sasaki, T.; Lynch, K.L.; Mueller, C.V.; Friedman, S.; Freitag, M.; Lewis, Z.A. Heterochromatin controls γH2A localization in Neurospora crassa. Eukaryot. Cell 2014, 13, 990–1000.
- Mastroeni, D.; Delvaux, E.; Nolz, J.; Tan, Y.; Grover, A.; Oddo, S.; Coleman, P.D. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3121–3129.
- Benhelli-Mokrani, H.; Mansuroglu, Z.; Chauderlier, A.; Albaud, B.; Gentien, D.; Sommer, S.; Schirmer, C.; Laqueuvre, L.; Josse, T.; Buée, L.; et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. 2018, 46, 11405–11422.
- Schindowski, K.; Bretteville, A.; Leroy, K.; Begard, S.; Brion, J.-P.; Hamdane, M.; Buee, L. Alzheimer’s disease-like tau ’neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2016, 169, 599–616.
- Hondius, D.C.; van Nierop, P.; Li, K.W.; Hoozemans, J.J.; van der Schors, R.C.; van Haastert, E.S.; van der Vies, S.M.; Rozemuller, A.J.; Smit, A.B. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 2016, 12, 654–668.
- Lane, D.P. Cancer. p53, Guardian of the genome. Nature 1992, 358, 15–16.
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311.
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132.
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000, 275, 29426–29432.
- Dawson, R.; Muller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141.
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246.
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.; Blagosklonny, M.; Fojo, T. P53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2000, 2, 709–717.
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Muñoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968.
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau protein disrupts Nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940.
- Diez, L.; Wegmann, S. Nuclear Transport Deficits in Tau-Related Neurodegenerative Diseases. Front. Neurol. 2020, 11, 1056.
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845.
- Montalbano, M.; McAllen, S.; Puangmalai, N.; Sengupta, U.; Bhatt, N.; Johnson, O.D.; Kharas, M.G.; Kayed, R. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat. Commun. 2020, 11, 4305.
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci. 2010, 33, 230–240.
- Bartolotti, N.; Bennett, D.A.; Lazarov, O. Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 2016, 21, 1158–1166.
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.-H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781.
- Mahoney, R.; Ochoa Thomas, E.; Ramirez, P.; Miller, H.E.; Beckmann, A.; Zuniga, G.; Dobrowolski, R.; Frost, B. Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium. Cell Rep. 2020, 32.
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714.
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534.
- Tracy, T.E.; Gan, L. Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 51, 134–138.
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. 2015, 35, 14234–14250.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39.
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce an Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6.
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J. Neurosci. 2018, 38, 10595–10606.
- Hill, E.; Karikari, T.K.; Moffat, K.G.; Richardson, M.J.E.; Wall, M.J. Introduction of Tau Oligomers into Cortical Neurons Alters Action Potential Dynamics and Disrupts Synaptic Transmission and Plasticity. eNeuro 2019, 6.
- Decker, J.M.; Krüger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol. Commun. 2015, 3, 23.
- Shrivastava, A.N.; Redeker, V.; Pieri, L.; Bousset, L.; Renner, M.; Madiona, K.; Mailhes-Hamon, C.; Coens, A.; Buée, L.; Hantraye, P.; et al. Clustering of tau fibrils impairs the synaptic composition of alpha3-Na+/K+-ATPase and AMPA receptors. EMBO J. 2019, 38, e99871.
- Regan, P.; Cho, K. The Role of Tau in the Post-synapse. Adv. Exp. Med. Biol. 2019, 1184, 113–121.

