 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Akeem Adeyemi Oladipo | -- | 1985 | 2026-07-14 09:49:29 | | | |
| 2 | Catherine Yang | Meta information modification | 1985 | 2026-07-15 02:15:23 | | |
Video Upload Options
Engineered nanomaterials (ENMs) deployed for aquatic remediation present a fundamental ecotoxicological paradox: the nanoscale physicochemical properties driving efficient pollutant degradation—specifically high specific surface energy, tunable redox potentials, and unsaturated coordination sites—simultaneously induce severe cytotoxicity in non-target aquatic organisms. The mechanistic origins of aquatic nanotoxicity stem from interfacial electron transfer, thermodynamics of oxidative dissolution, and colloidal destabilization, which collectively initiate cellular damage via uncontrolled reactive oxygen species (ROS) generation and the endocytic "Trojan Horse" pathway. In natural waters, environmental transformations such as dissolved organic matter adsorption form a dynamic eco-corona, fundamentally altering colloidal stability, dissolution kinetics, and bioavailability. Resolving this duality requires transitioning from reactive post hoc risk assessments to a proactive Safe-and-Sustainable-by-Design (SSbD) framework, utilizing electronic band-gap engineering, heteroatom doping, and core-shell mesoporous architectures to systematically decouple remediation efficacy from ecological hazard.
1. Introduction to the Nanoremediation Paradox
The application of engineered nanomaterials (ENMs) for the remediation of contaminated aquatic systems represents a major paradigm shift in environmental engineering. Nanoscale remediation agents—including zero-valent iron (nZVI), titanium dioxide (TiO2), zinc oxide (ZnO), and functionalized metal-organic framework (MOF) composites—demonstrate exceptionally high efficacy in degrading recalcitrant organic pollutants, sequestering heavy metal ions, and inactivating pathogenic microorganisms through advanced oxidation processes (AOPs) and high-capacity adsorption isotherms.[1]
Deploying ENMs in open aquatic ecosystems presents a fundamental scientific duality: the specific physicochemical properties that govern remedial functionality—namely ultra-high specific surface area, elevated surface reactivity, and engineered electronic band structures—are the exact shared origins of ecotoxicological hazard.[1][2][3] While a highly reactive photocatalytic nanoparticle may achieve >90% mineralization of a target pharmaceutical or pesticide contaminant, that same non-selective interfacial reactivity can indiscriminately disrupt the cellular architecture of non-target aquatic organisms. Across trophic levels, exposure manifests as inhibition of photosynthetic electron transport in microalgae (Chlamydomonas reinhardtii), reproductive impairment and immobilization in pelagic filter-feeders (Daphnia magna), and gill epithelial ulceration, ion-regulatory failure, and systemic oxidative stress in teleost fish (Danio rerio).[2][3] Decoupling environmental remediation efficacy from ecotoxicological hazard is the central engineering challenge of modern environmental nanotechnology.
2. Physicochemical Drivers of Aquatic Toxicity
The ecotoxicological profile of an ENM in an aquatic system is governed by its nanoscale physical state and interfacial thermodynamics rather than its bulk chemical identity.[4]
2.1. Surface Energy and Size-Dependent Scaling
As particle diameter (d) decreases below 100 nm, the ratio of surface atoms (Ns) to total atoms (Nt) scales inversely with size according to the geometric approximation:
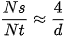
This rapid geometric scaling generates a high surface density of unsaturated coordination sites, atomic step edges, and surface vacancies.[4] The resulting excess surface free energy (ΔGsurface) lowers the thermodynamic activation barrier for interfacial electron transfer and substrate adsorption. Consequently, biological reactivity accelerates significantly, allowing sub-micron ENMs to bypass physiological barriers—such as the teleost chorion or algal cellulose cell walls—more readily than bulk equivalents.[4]
2.2. Surface Charge and Colloidal Stability
The electrical double layer surrounding an aqueous ENM is characterized by its zeta potential (ζ). Cationic ENMs exhibiting zeta potentials exceeding +20 mV demonstrate significantly higher cytotoxicity than anionic or neutral counterparts.[5] This enhanced toxicity is driven by strong electrostatic attraction to the negatively charged phospholipid bilayers of biological membranes, which induces membrane depolarization, enhanced ion permeability, and physical pore formation.
Colloidal stability and aggregation kinetics in aquatic media are dictated by Derjaguin-Landau-Verwey-Overbeek (DLVO) theory, where the total potential energy of interaction (VT) is the sum of attractive van der Waals forces (VvdW) and repulsive electrostatic double-layer forces (VEDL):
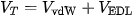
In high-ionic-strength aquatic environments (such as marine, estuarine, or hard freshwater systems), double-layer compression reduces the Debye screening length, significantly diminishing VEDL.[5] This reduction eliminates the energy barrier to aggregation, causing rapid homoaggregation and subsequent sedimentation into the benthic zone. This physical transformation shifts the primary ecological exposure hazard from pelagic filter-feeders in the water column to benthic detritivores and sediment-dwelling organisms.[5]
2.3. Thermodynamics of Oxidative Dissolution
For metal and metal-oxide ENMs (e.g., Ag, ZnO, CuO), acute aquatic toxicity is governed primarily by oxidative dissolution kinetics rather than the physical presence of the solid nanoparticle.[6] The equilibrium dissolution of silver nanoparticles (AgNPs) in oxygenated aqueous media proceeds via the coupled redox reaction:
The liberated free aqueous metal cations (Ag+, Zn2+, Cu2+) act as potent soft acids under Pearson’s Hard and Soft Acids and Bases (HSAB) principle. These cations bind with high affinity to soft base nucleophiles, particularly protein sulfhydryl (-SH) groups on cysteine residues. This complexation inactivates critical enzymatic cascades, disrupts mitochondrial respiration, and inhibits Na+/K+-ATPase and Ca2+-ATPase ion-regulation pumps within teleost gill epithelia.[6]
3. Molecular Initiating Events (MIEs) and Cellular Internalization
Within mechanistic ecotoxicology, nanomaterial hazard is structured through the Adverse Outcome Pathway (AOP) framework. The cascade begins with a Molecular Initiating Event (MIE)—the initial direct physical or chemical interaction between the ENM surface and a biological macromolecule (lipid, protein, or nucleic acid).[7]
3.1. Internalization Kinetics and the "Trojan Horse" Mechanism
In aquatic organisms, ENMs enter systemic circulation through branchial gill filtration, enteral ingestion via the alimentary canal, or dermal absorption. At the cellular level, internalization occurs predominantly via endocytic pathways, including clathrin-mediated endocytosis, caveolae-dependent endocytosis, and macropinocytosis.
Once internalized, ENMs are trafficked to early endosomes, which systematically mature into late endosomes and fuse with lysosomes to form phagolysosomes. This intracellular routing activates the "Trojan Horse" dissolution mechanism:[6][7]
-
Relatively insoluble ENMs are engulfed extracellularly and compartmentalized within highly acidic lysosomes (pH 4.5–5.0).
-
The proton-rich environment alters the Nernstian redox equilibrium, accelerating the thermodynamics of metal dissolution:
$$E = E^0 - \frac{RT}{nF} \ln Q$$ -
This intracellular dissolution generates an intense, localized burst of free metal cations directly within the lysosomal lumen.
-
The high cation concentration overwhelms local metallothionein buffering capacities, inducing lipid peroxidation of the lysosomal membrane.
-
The resulting lysosomal membrane permeabilization (LMP) spills hydrolytic digestive proteases (such as cathepsins B and D) and toxic metal ions directly into the cytosol, initiating mitochondrial depolarization, caspase-3 activation, and programmed cell death (apoptosis or necrosis).[7]
4. Oxidative Stress and ROS-Mediated Pathways
The most universally conserved mechanism of nanotoxicity across aquatic taxa is the induction of intracellular oxidative stress driven by the overproduction of Reactive Oxygen Species (ROS). While baseline ROS generation is a regulated byproduct of mitochondrial electron transport, redox-active ENMs catalyze an uncontrolled intracellular burst, frequently elevating physiological radical concentrations by 1.5- to 2.0-fold.[2][3][7]
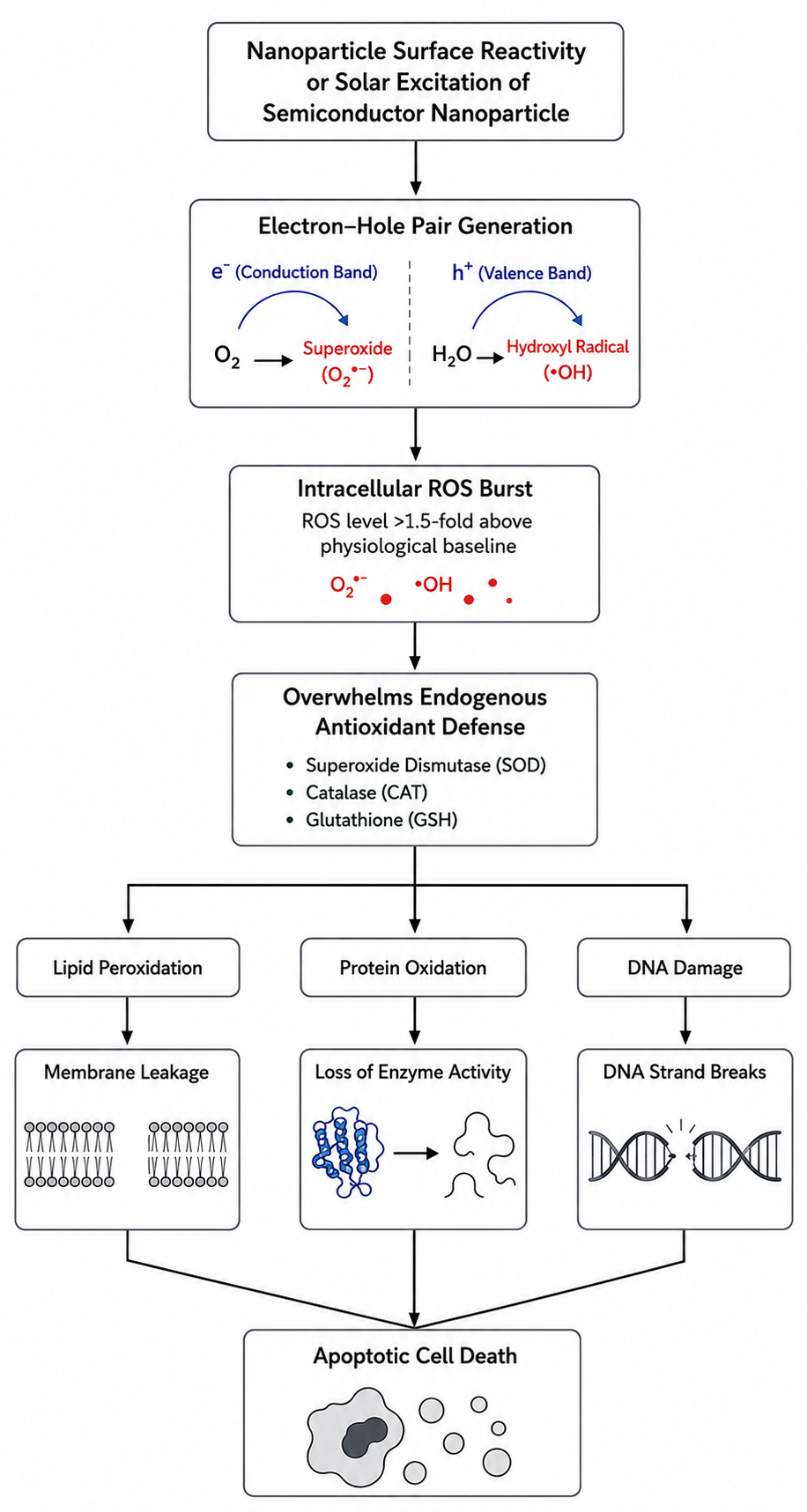
4.1. Mechanisms of Radical Generation
For semiconductor ENMs (e.g., TiO2, ZnO), band-gap excitation by ambient solar or ultraviolet radiation promotes an electron from the valence band (VB) to the conduction band (CB), generating an active electron-hole pair:
When the conduction band potential is sufficiently negative relative to the redox potential of adsorbed oxygen, the photo-excited electron reduces molecular oxygen to superoxide radicals (O2•-):
Concurrently, positive valence band holes oxidize interfacial water molecules or hydroxide ions to generate hydroxyl radicals (OH•), representing one of the most electrophilic and reactive oxidants in biological systems:[8]
For transition-metal-based ENMs containing iron, copper, or manganese, dissolved cations catalyze intracellular Fenton and Haber-Weiss cycles within the cytosol and mitochondria:
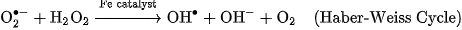
4.2. Biochemical Downstream Consequences
When ROS production outpaces endogenous antioxidant buffering systems—specifically superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx), along with depletion of reduced glutathione (GSH)—the cell experiences severe oxidative stress, resulting in three convergent pathways of macromolecular damage:[3][7]
-
Lipid Peroxidation: Hydroxyl radicals abstract hydrogen atoms from polyunsaturated fatty acids (PUFAs) within phospholipid membranes. This abstraction initiates a self-propagating chain reaction producing lipid hydroperoxides (LOOH) and cytotoxic aldehydes, specifically malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). This structural degradation impairs membrane fluidity, collapses ion electrochemical gradients, and causes cytolysis.
-
Protein Oxidation: Direct radical attack induces carbonyl formation and disulfide bond cross-linking at catalytic amino acid residues (particularly methionine and cysteine), resulting in protein misfolding, aggregation, and loss of enzymatic function.
-
Genotoxicity: Hydroxyl radicals penetrate the nuclear envelope, generating oxidative DNA adducts such as 8-hydroxy-2'-deoxyguanosine (8-OHdG), initiating single- and double-strand breaks, and inducing chromosomal aberrations. Persistent genotoxic damage upregulates p53 transcription pathways, arresting the cell cycle and inducing apoptotic cascades.[7]
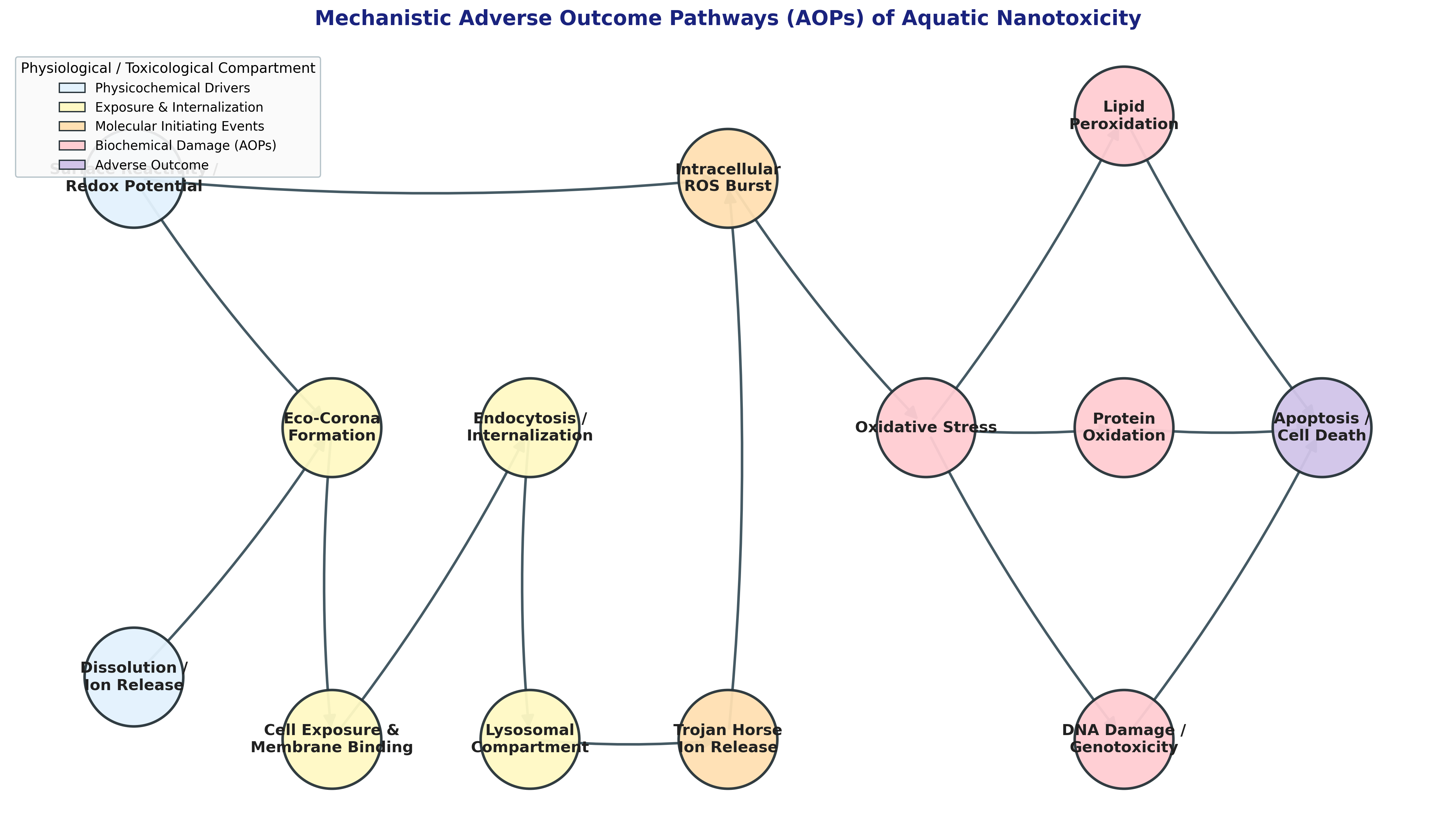
5. Environmental Transformations: The Eco-Corona Effect
Evaluating ENM ecotoxicity exclusively using pristine particles synthesized in laboratory water represents a significant methodological limitation. Upon discharge into natural aquatic ecosystems, the interfacial identity of an ENM is immediately transformed by its surrounding hydrochemical matrix.[3][5][9]
5.1. Thermodynamics of Eco-Corona Formation
Natural waters contain high concentrations of Dissolved Organic Matter (DOM), comprising humic acids, fulvic acids, and microbially derived extracellular polymeric substances (EPS). These biological macromolecules rapidly adsorb onto the high-energy nanoparticle surface via ligand exchange, hydrophobic interactions, and hydrogen bonding. This adsorption results in a structurally dynamic, layered organic coating termed the eco-corona.[3][9]
The formation of the eco-corona alters the nanomaterial's biological identity, kinetics, and environmental fate through several distinct mechanisms:
-
Surface Passivation: A tightly bound, dense organic layer (the "hard corona") acts as a steric physical barrier. This barrier blocks surface catalytic sites, inhibits oxidative dissolution kinetics by limiting proton accessibility, and quenches interfacial ROS generation, frequently reducing acute contact cytotoxicity.
-
Colloidal Destabilization and Bridging Flocculation: In the presence of divalent natural cations (Ca2+, Mg2+), carboxylate functional groups within adsorbed humic substances undergo intermolecular bridging. This bridging neutralizes surface charge and accelerates heteroaggregation and sedimentation, clearing the pelagic water column of nanoparticles but significantly concentrating the ecotoxicological load within benthic sediments.[5]
-
The "Trojan Corona" Uptake Pathway: Conversely, specific EPS and protein coatings can facilitate receptor-mediated endocytosis. By masking the inorganic core with endogenous biomolecules, the eco-corona can mimic natural nutrients or signaling ligands, paradoxically enhancing cellular uptake rates in filter-feeding organisms despite passivating the reactive inorganic surface.[3][7][6][9]
6. The Paradigm Shift: Safe-and-Sustainable-by-Design (SSbD)
Historically, environmental risk assessment of nanomaterials has operated post hoc—prioritizing the synthesis and optimization of an ENM for remediation efficacy first, and evaluating ecotoxicity only as a secondary prerequisite prior to deployment. This sequential approach repeatedly results in the regulatory restriction or complete abandonment of highly effective remediation agents due to unforeseen aquatic toxicity.
The modern engineering paradigm relies on Safe-and-Sustainable-by-Design (SSbD) principles, which embed mechanistic toxicology as an a priori design parameter during advanced materials synthesis. By establishing the shared electronic, structural, and thermodynamic origins of reactivity and toxicity, materials scientists can systematically manipulate nanoscale architectures to decouple remediation efficacy from ecological hazard.[3][9]
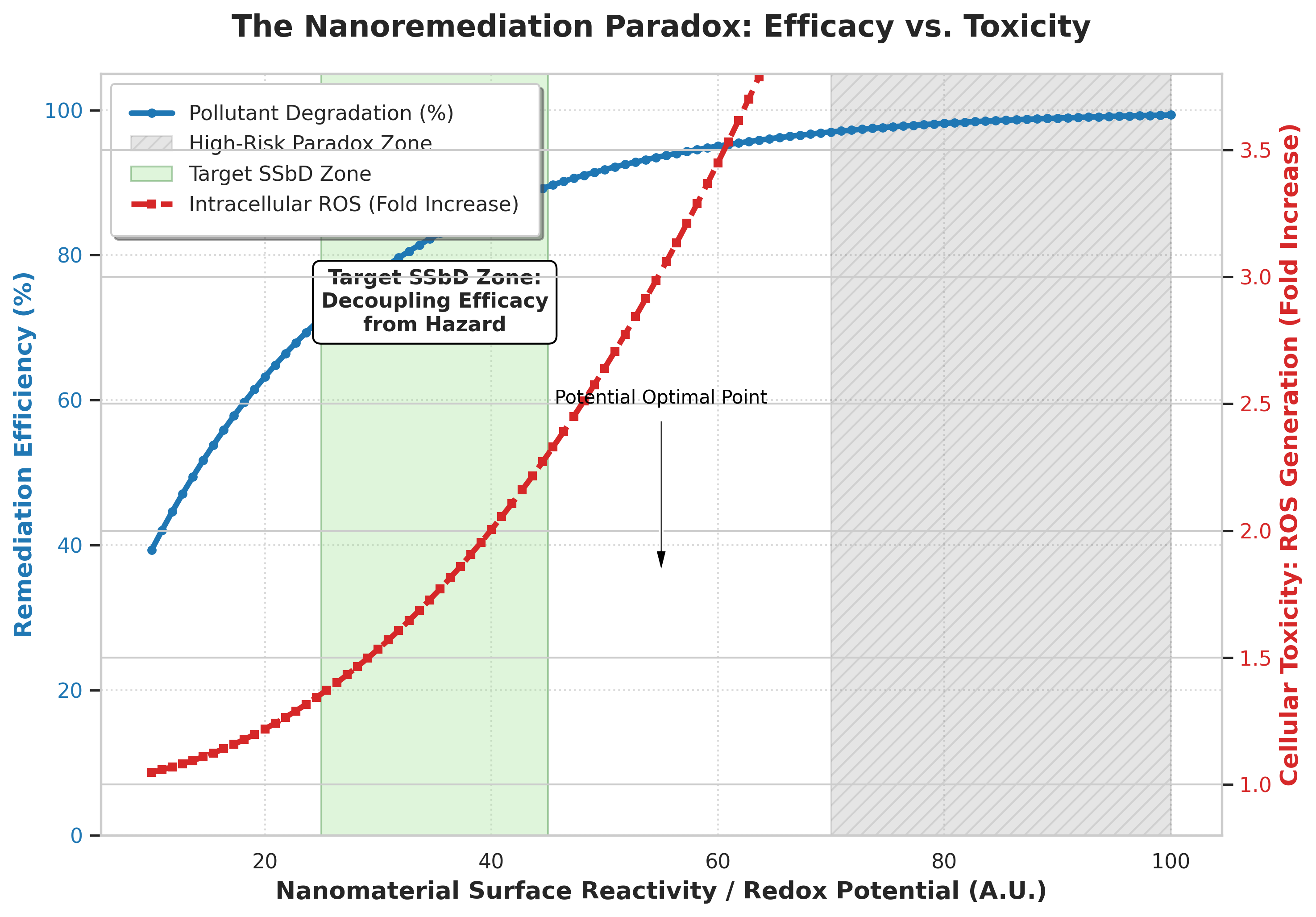
6.1. Rational Engineering Strategies
Electronic Band-Gap and Defect Engineering
By doping semiconductor photocatalysts with specific non-metal heteroatoms (such as nitrogen, sulfur, or carbon) or controlling crystal lattice oxygen vacancies, the electronic band structure can be altered to shift radical selectivity.[8] For example, modifying the conduction band edge position can thermodynamically favor the reduction of dissolved oxygen to singlet oxygen (1O2) over the generation of hydroxyl radicals (OH•). While 1O2 remains highly effective at degrading electron-rich organic pollutants (such as phenols and pharmaceuticals), its shorter half-life in aqueous media (~3.5 µs) and lower redox potential prevent it from initiating widespread lipid peroxidation across biological membranes, drastically lowering acute cytotoxicity.[9]
Core-Shell and Mesoporous Size-Exclusion Architectures
Encapsulating redox-active or soluble metal cores (e.g., nZVI, Ag, ZnO) within chemically inert, biocompatible mesoporous shells (such as amorphous silica, mSiO2, or titanium-based metal-organic frameworks) provides physical and kinetic isolation. This architecture operates on strict size-exclusion kinetics:
-
Low-molecular-weight target organic contaminants (0.5–1.5 nm) diffuse freely through the engineered mesopores (2–5 nm pore diameter) to access the reactive catalytic core.
-
High-molecular-weight biological entities—such as phospholipid membranes, cellular organelles, and functional extracellular enzymes (>5 nm)—are physically excluded from direct contact with the catalytic surface.
This spatial compartmentalization prevents contact-dependent ROS degradation of cell membranes and inhibits the Trojan Horse dissolution pathway, successfully achieving high-efficiency aquatic remediation without triggering ecotoxicological events in non-target species.[1][3][7][9]
References
- Guerra, F.D.; Attia, M.F.; Whitehead, D.C.; Alexis, F. Nanotechnology for environmental remediation: Materials and applications. Molecules. 2018, 23, 1760.
- Bundschuh, M.; Filser, J.; Wagner, S.; Klingelhöfer, D.; Sander, M.; Schäfer, R.B. Nanoparticles in the environment: Where do we come from, where do we go to?. Environ. Sci. Eur.. 2018, 30, 6.
- Oladipo, A.A. The double-edged nanoparticle: remediation benefits vs. mechanistic toxicity risks in aquatic systems. Environ. Sci.: Nano . 2026, 13 (1), 79–105.
- Auffan, M.; Rose, J.; Bottero, J.Y.; Lowry, G.V.; Jolivet, J.P.; Wiesner, M.R. Towards a definition of inorganic nanoparticles from an environmental, health and safety perspective. Nat. Nanotechnol.. 2009, 4, 634–641.
- Lowry, G.V.; Gregory, K.B.; Apte, S.C.; Lead, J.R. Transformations of nanomaterials in the environment. Environ. Sci. Technol.. 2012, 46, 6893–6899.
- Notter, D.A.; Mitrano, D.M.; Nowack, B. Are nanosized or dissolved metals more toxic in the environment?. Environ. Toxicol. Chem.. 2018, 37, 2150–2159.
- Fadeel, B.; Farcal, L.; Hardy, B.; Vázquez-Campos, S.; Hristozov, D.; Marcomini, A.; Lynch, I. Advanced tools for the safety assessment of nanomaterials.. Nat. Nanotechnol.. 2018, 13, 537–543.
- Oladipo, A.A. Spinel Ferrite–Based Advanced Oxidation Processes for Recalcitrant and Emerging Contaminants: Mechanistic Insights, Structure–Property Relationships, and Hybrid AOP Strategies. Journal of Environmental Chemical Engineering . 2026, 14, 122234.
- Lynch, I.; Weiss, C.; Valsami-Jones, E. A strategy for predicting the impacts of nanomaterials on biomolecules, cells and tissues.. Nano Today. 2014, 9, 266–270.


