 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hideaki Kaneto | + 1773 word(s) | 1773 | 2020-12-29 08:49:51 | | | |
| 2 | Catherine Yang | Meta information modification | 1773 | 2020-12-30 05:10:48 | | |
Video Upload Options
Under healthy conditions, pancreatic β-cells produce and secrete the insulin hormone in response to blood glucose levels. Under diabetic conditions, however, β-cells are compelled to continuously secrete larger amounts of insulin to reduce blood glucose levels, and thereby, the β-cell function is debilitated in the long run.
1. MafA and PDX-1 Play a Crucial Role in Pancreatic β-Cells
Under healthy conditions, pancreatic β-cells function to produce and secrete the insulin hormone in response to high glucose concentrations. Under diabetic conditions, however, β-cells are compelled to secrete larger amounts of insulin continuously in order to reduce blood glucose levels. Such succession is some grueling work for β-cells themselves, and thereby, the β-cell function is debilitated in the long run (so called “pancreatic β-cell glucose toxicity”) [1][2][3]. Chronic hyperglycemia reduces insulin biosynthesis and secretion together with reduced expression of insulin gene transcription factors such as MafA and PDX-1. In clinical practice, it is very important to alleviate such β-cell glucose toxicity so that the aggravation of diabetes mellitus is forestalled. In addition, insulin signaling in insulin target tissues (liver, skeletal muscle, and adipose tissue) is weakened by the burden of glucose toxicity, leading to the development of insulin resistance. Such debilitation of the β-cell function and development of insulin resistance lead to further aggravation of type 2 diabetes mellitus (Figure 1).
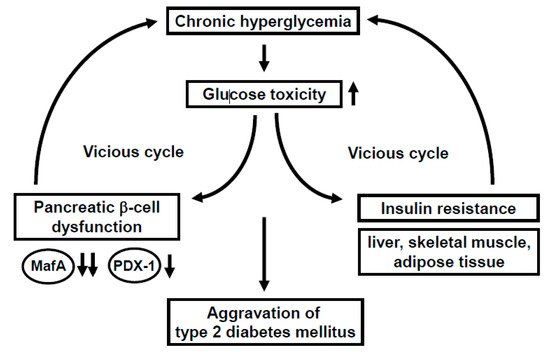
Figure 1. Involvement of glucose toxicity in pancreatic β-cell dysfunction and insulin resistance. Under diabetic conditions, pancreatic β-cell function is gradually debilitated by the burden of glucose toxicity. In addition, insulin signaling in insulin target tissues (liver, skeletal muscle, and adipose tissue) is weakened by the burden of glucose toxicity, leading to the development of insulin resistance. Such debilitation of the β-cell function and development of insulin resistance bring about a vicious cycle and finally lead to further aggravation of type 2 diabetes mellitus.
MafA is a strong transcription factor for the insulin gene [4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19]. In MafA knockout mice, insulin biosynthesis and secretion are reduced, leading to diabetes mellitus, indicating the importance of MafA in β-cells [8]. The MafA expression level is markedly reduced in a diabetic state [11], but it is preserved after alleviation of glucose toxicity with some anti-diabetic agent [16][17][18][19]. Furthermore, we reported that in β-cell-specific and conditional (tamoxifen-induced) MafA overexpressing transgenic db/db mice, serum insulin levels were higher and blood glucose levels were lower compared to their littermates. In addition, the β-cell mass was preserved in MafA overexpressing db/db mice mainly due to the reduction of apoptotic cell death. We think that these findings clearly underscore the importance of MafA in β-cells and become unequivocal evidence that downregulation of MafA expression is associated with β-cell glucose toxicity (Figure 1).
PDX-1, another important transcription factor for the insulin gene, plays a crucial role in pancreas development and β-cell differentiation and maintenance of mature β-cell function [20][21][22][23][24][25][26][27][28][29][30][31]. In PDX-1 knockout mice, pancreas formation is not observed at all [23]. In mature β-cells, PDX-1 transactivates several genes including insulin, glucokinase, and glut2. However, the expression level of PDX-1 is reduced in a diabetic state [26][29], which we think is associated with β-cell failure observed in diabetes. Indeed, we recently reported that β-cell-specific and conditional (tamoxifen-induced) PDX-1 overexpressing transgenic Akita mice showed lower blood glucose levels and lower HbA1C values compared to their littermates, accompanied by increased insulin secretion after glucose loading [31]. Expression levels of MafA and glucokinase were preserved in PDX-1 overexpressing Akita mice. We think that such new findings suggest that the downregulation of PDX-1 expression found in diabetes undermines insulin biosynthesis and secretion which explains, at least in part, the mechanism for β-cell glucose toxicity (Figure 1).
2. Incretin Signaling Plays an Important Role in Pancreatic β-Cells
In response to the ingestion of food, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are released from the gastrointestinal tract both of which stimulate insulin secretion. GLP-1 and GIP bind to the GLP-1 and GIP receptor in β-cells, respectively, which increases intracellular cAMP levels. Increased cAMP levels lead to augmenting insulin secretion, reducing β-cell apoptosis, and facilitating β-cell proliferation (Figure 2). Incretin-related drugs such as glucagon-like peptide-1 receptor (GLP-1R) activators or dipeptidyl peptidase-IV (DPP-IV) inhibitors are known to ameliorate glycemic control and reduce the progression of β-cell dysfunction in human subjects, as well as rodent models [32][33][34][35]. Indeed, it was reported that the GLP-1 receptor activator liraglutide preserved pancreatic β-cell function through the reduction of glucose toxicity, as well as through its direct effect on β-cells [32][35]. The DPP-IV inhibitor suppresses the activity of DPP-IV, which is a splitting enzyme of incretin and increases serum levels of GLP-1 and GIP. Both incretins stimulate insulin secretion in a glucose-dependent manner and GLP-1 suppresses glucagon secretion. Indeed, it has been reported that DPP-IV inhibitors preserve pancreatic β-cell function through the reduction of glucose toxicity, as well as through the direct protective effect of incretin [33][34].
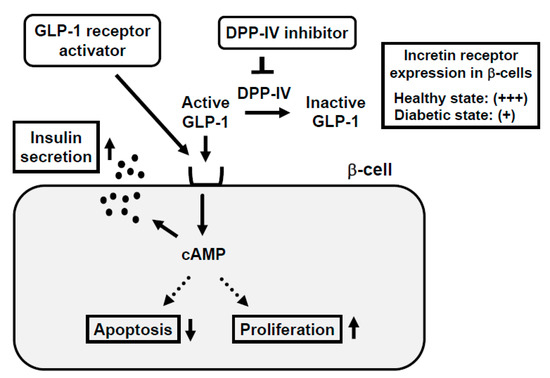
Figure 2. Mechanism of action of incretin and incretin-based agents in pancreatic β-cells and reduced expression of incretin receptor in β-cells in a diabetic state. Binding of incretin to incretin receptor in β-cell membrane increases intracellular cAMP levels, which finally leads to augmentation of insulin secretion from β-cells, reduction of β-cell apoptosis, and facilitation of β-cell proliferation. In a healthy state, the incretin receptor is abundantly expressed in the β-cell membrane, but in a diabetic state, the incretin receptor expression is substantially reduced.
3. Incretin Receptor Expression in β-Cells Is Downregulated under Diabetic Conditions: Incretin-Based Agents Exert More Protective Effects on β-Cells at an Early Stage of Diabetes rather than an Advanced Stage
It is well known that the incretin hormone action is significantly reduced in subjects with type 2 diabetes mellitus. Moreover, it has been reported that expression levels of incretin receptors are decreased in a diabetic state, which is likely involved in the impaired incretin effects and the progression of β-cell failure found in diabetes [16][36][37][38](Figure 2). In addition, it has been suggested that the decreased expression of transcription factor 7-like 2 (TCF7L2), which is a transcription factor of incretin receptors and plays a crucial role in the maintenance of β-cell function, is involved in the downregulation of the incretin receptor expression in β-cells found in diabetes [39] [40][41].
It was reported that incretin-related drug liraglutide increased the β-cell function and mass at an early stage of diabetes, but these effects were attenuated at an advanced stage [35]. Only at an early stage, insulin biosynthesis and glucose-stimulated insulin secretion were markedly increased by liraglutide [35]. In addition, only at an early stage, expression levels of various insulin gene transcription factors such as MafA and PDX-1 were upregulated by liraglutide. We think that the increased expression of such factors at an early stage explains the increased insulin biosynthesis and secretion observed at an early stage. It is likely that the recovery of MafA expression is particularly important for the recovery of β-cell function and amelioration of glycemic control, since MafA regulates not only the insulin gene but also various factors related to the glucose-stimulated insulin secretion. In addition, the expression of GLP-1 receptor was reduced at an advanced stage, which we think explains the reason why liraglutide did not exert beneficial effects at an advanced stage compared to an early stage [35]. Taken together, we think that it is very important to start using incretin-based drugs at an early stage of diabetes to enable incretin-based drugs to fully exert their effect for the protection of β-cell function.
4. Impaired Insulin Signaling in Endothelial Cells Leads to Pancreatic β-Cell Dysfunction
In general, the main insulin target tissues are the liver, skeletal muscle, and adipose tissues, but there are many kinds of tissues and/or cells in which insulin signaling plays some important role. In endothelial cells, binding of insulin to insulin receptor on its cell surface activates insulin receptor substrate (IRS), phosphoinositide 3-kinase (PI3K), and 3-phosphoinositide-dependent protein kinase-1 (PDK1). Such activated insulin signaling leads to augment nitric oxide production in endothelial cells. Indeed, there have been several reports showing the importance of insulin signaling in endothelial cells [42][43][44][45][46]. Since it is known that the endothelial cell dysfunction is observed under diabetic conditions, it is possible that such endothelial dysfunction brings out hypoxia and ischemia in various tissues through insufficiency of nitric oxide production. In addition, it is known that pancreatic islets are particularly vulnerable to various stimuli including hypoxia and ischemia, and thereby it is also possible that endothelial dysfunction leads to exacerbation of pancreatic β-cell function.
Recently, we examined the possible role of PDK1, one of the important molecules in insulin signaling in vascular endothelial cells, in the maintenance of pancreatic β-cell mass and function. As a result, vascular endothelial-specific PDK1 knockout mice presented reduced β-cell mass and impaired β-cell function [47] (Figure 3). These mice also presented reduced blood flow of pancreas and/or islets and hypoxia of β-cells. In these KO mice, the β-cell mass was significantly reduced and the blood vessel region in islets was significantly decreased. In addition, in these KO mice, incretin secretion was augmented after the oral glucose tolerance test but insulin secretion was impaired, suggesting the impairment of incretin sensitivity in islets of KO mice. Insulin, MafA, PDX-1, GLP-1, and GIP receptor expression levels were all significantly decreased in islets of KO mice [47]. The microsphere experiment elucidated the remarkably reduced islet blood flow, and HIF1α and its downstream factor expression levels were significantly increased in islets of KO mice, indicating that islets of KO mice were in a more hypoxic state compared to the control mice. Consequently, ER stress-related gene expression levels were significantly elevated and inflammatory cytokine levels were increased in islets of KO mice [47].

Figure 3. Association of endothelial cell dysfunction with pancreatic β-cell dysfunction: Involvement of hypoxia, provoked endoplasmic reticulum (ER) stress, and inflammatory reaction. Ablation of endothelial PDK1 reduces vascularity in islets, and both pancreatic and islet blood flow are decreased, which lead to hypoxia in islets and induction of ER stress and inflammation. Therefore, it is likely that vascular endothelial PDK1 plays an important role in the maintenance of pancreatic β-cell mass and function by maintaining the vascularity of the pancreas and islets and protecting them from hypoxia, ER stress, and inflammation.
Taken together, ablation of endothelial PDK1 reduces vascularity in islets, and both pancreatic and islet blood flow are decreased, which lead to hypoxia in islets and induction of ER stress and inflammation. Therefore, it is likely that vascular endothelial PDK1 plays an important role in the maintenance of pancreatic β-cell mass and function by maintaining the vascularity of the pancreas and islets and protecting them from hypoxia, hypoxia-related ER stress, and inflammation. These are novel concepts to explain the underlying molecular mechanism for pancreatic β-cell failure and we think that such findings would be useful when we think about future strategies for type 2 diabetes mellitus (Figure 3).
References
- Rhodes, C.J. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384.
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758.
- Kaneto, H. Pancreatic β-cell glucose toxicity in type 2 diabetes mellitus. Curr. Diabetes Rev. 2015, 11, 2–6.
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742.
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49,903–49,910.
- Matsuoka, T.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol. Cell. Biol. 2003, 23, 6049–6062.
- Kaneto, H.; Matsuoka, T.; Nakatani, Y.; Miyatsuka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. A crucial role of MafA as a novel therapeutic target for diabetes. J. Biol. Chem. 2005, 280, 15047–15052.
- Zhang, C.; Moriguchi, T.; Kajihara. M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; Kudo, T.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976.
- Matsuoka, T.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet β cell function. Mol. Endocrinol. 2007, 21, 2764–2774.
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358.
- Matsuoka, T.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic b-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720.
- Yamamoto, K.; Matsuoka, T.; Kawashima, S.; Takebe, S.; Kubo, N.; Miyatsuka, T.; Kaneto, H.; Shimomura, I. A novel function of Onecut 1 as a negative regulator of MafA. J. Biol. Chem. 2013, 288, 21,648–21,658.
- Matsuoka, T.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving MafA expression in diabetic islet β-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657.
- Kaneto, H.; Matsuoka, T. Role of pancreatic transcription factors in maintenance of mature β-cell function. Int. J. Mol. Sci. 2015, 16, 6281–6297.
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574.
- Kawashima, S.; Matsuoka, T.; Kaneto, H.; Tochino, Y.; Kato, K.; Yamamoto, K.; Yamamoto, T.; Matsuhisa, M.; Shimomura, I. Effect of alogliptin, pioglitazone and glargine on pancreatic β-cells in diabetic db/db mice. Biochem. Biophys. Res. Commun. 2011, 404, 534–540.
- Shimo, N.; Matsuoka, T.; Miyatsuka, T.; Takebe, S.; Tochino, Y.; Takahara, M.; Kaneto, H.; Shimomura, I. Short-term selective alleviation of glucotoxicity and lipotoxicity ameliorates the suppressed expression of key β-cell factors under diabetic conditions. Biochem. Biophys. Res. Commun. 2015, 467, 948–954.
- Okauchi, S.; Shimoda, M.; Obata, A.; Kimura, T.; Hirukawa, H.; Kohara, K.; Mune, T.; Kaku, K.; Kaneto, H. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic β-cells in obese type 2 diabetic db/db mice. Biochem. Biophys. Res. Commun. 2016, 470, 772–782.
- Kimura, T.; Obata, A.; Shimoda, M.; Okauchi, S.; Kanda-Kimura, Y.; Nogami, Y.; Hirukawa, H.; Kohara, K.; Nakanishi, S.; Mune, T.; et al. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic β-cells in obese diabetic db/db mice: The earlier and longer, the better. Diabetes Obes. Metab. 2018, 20, 2442–2457.
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing-transactivator of the insulin gene. EMBO. J. 1993, 12, 4251–4259.
- Leonard, J.; Peers, B.; Johnson, T.; Ferreri, K.; Lee, S.; Montminy, M.R. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol. Endocrinol. 1993, 7, 1275–1283.
- Miller, C.P.; McGehee, R.E.; Habener, J.F. IDX-1: A new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO. J. 1994, 13, 1145–1156.
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 37, 606–609.
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. β-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the b-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768.
- Holland, A.M.; Hale, M.A.; Kagami, H.; Hammer, R.E.; MacDonald, R.J. Experimental control of pancreatic development and maintenance. Proc. Natl. Acad. Sci. USA 2002, 99, 12236–12241.
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018.
- Noguchi, H.; Kaneto, H.; Weir, G.C.; Bonner-Weir, S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003, 52, 1732–1737.
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes 2005, 54, 1009–1022.
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098.
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632.
- Yamamoto, Y.; Miyatsuka, T.; Sasaki, S.; Miyashita, K.; Kubo, N.; Shimo, N.; Takebe, S.; Watada, H.; Kaneto, H.; Matsuoka, T.; et al. Recovered expression of Pdx1 improves β-cell failure in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 483, 418–424.
- Shimoda, M.; Kanda, Y.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Matsuki, M.; Kaku, K. The human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells via regulation of cell kinetics and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetologia 2011, 54, 10598–11108.
- Hamamoto, S.; Kanda, Y.; Shimoda, M.; Tatsumi, F.; Kohara, K.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Vildagliptin preserves the mass and function of pancreatic beta cells via the developmental regulation and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetes Obes. Metab. 2013, 15, 153–163.
- Hirukawa, H.; Kaneto, H.; Shimoda, M.; Kimura, T.; Okauchi, S.; Obata, A.; Kohara, K.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; et al. Combination of DPP-4 inhibitor and PPARγ agonist exerts protective effects on pancreatic β-cells in diabetic db/db mice through the augmentation of IRS-2 expression. Mol. Cell. Endocrinol. 2015, 413, 49–60.
- Kimura, T.; Kaneto, H.; Shimoda, M.; Hirukawa, H.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Protective effects of pioglitazone and/or liraglutide on pancreatic β-cells: Comparison of their effects between in an early and advanced stage of diabetes. Mol. Cell. Endocrinol. 2015, 400, 78–89.
- Xu, G.; Kaneto, H.; Laybutt, D.R.; Duvivier-Kali, V.; Trivedi, N.; Suzuma, K.; King, G.L.; Weir, G.C.; Bonner-Weir, S. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to the impaired incretin effects in diabetes. Diabetes 2007, 56, 1551–1558.
- Kubo, F.; Miyatsuka, T.; Sasaki, S.; Takahara, M.; Yamamoto, Y.; Shimo, N.; Watada, H.; Kaneto, H.; Gannon, M.; Matsuoka, T.; et al. Sustained expression of GLP-1 receptor differentially modulates β-cell functions in diabetic and nondiabetic mice. Biochem. Biophys. Res. Commun. 2016, 471, 68–74.
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399.
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735.
- Takamoto, I.; Kubota, N.; Nakaya, K.; Kumagai, K.; Hashimoto, S.; Kubota, T.; Inoue, M.; Kajiwara, E.; Katsuyama, H.; Obata, A.; et al. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia 2014, 57, 542–553.
- Mitchell, R.K.; Mondragon, A.; Chen, L.; McGinty, J.A.; French, P.M.; Ferrer, J.; Thorens, B.; Hodson, D.J.; Rutter, G.A.; Xavier, G.D. Selective disruption of Tcf7l2 in the pancreatic β cell impairs secretory function and lowers β cell mass. Hum. Mol. Genet. 2015, 24, 1390–1399.
- Kondo, T.; Vicent, D.; Suzuma, K. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J. Clin. Invest. 2003, 111, 1835–1842.
- Mukai, Y.; Rikitake, Y.; Shiojima, I.; Wolfrum, S.; Satoh, M.; Takeshita, K.; Hiroi, Y.; Salomone, S.; Kim, H.H.; Benjamin, L.E.; et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J. Clin. Invest. 2006, 116, 334–343.
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487.
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307.
- Hashimoto, S.; Kubota, N.; Sato, H.; Sasaki, M.; Takamoto, I.; Kubota, T.; Nakaya, K.; Noda, M.; Ueki, K.; Kadowaki, T. Insulin receptor substrate-2 (Irs2) in endothelial cells plays a crucial role in insulin secretion. Diabetes 2015, 64, 876–886.
- 47. Obata, A.; Kimura, T.; Obata, Y.; Shimoda, M.; Kinoshita, T.; Kohara, K.; Okauchi, S.; Hirukawa, H.; Kamei, S.; Nakanishi, S.; et al. Vascular endothelial PDK1 plays pivotal roles for maintenance of pancreatic beta-cell mass and function in adult male mice. Diabetologia 2019, 62, 1225–1236.


