Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gaetano Vattemi | -- | 2660 | 2024-03-12 07:12:38 | | | |
| 2 | Catherine Yang | Meta information modification | 2660 | 2024-03-12 07:22:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Guglielmi, V.; Cheli, M.; Tonin, P.; Vattemi, G. Sporadic Inclusion Body Myositis. Encyclopedia. Available online: https://encyclopedia.pub/entry/56144 (accessed on 24 July 2026).
Guglielmi V, Cheli M, Tonin P, Vattemi G. Sporadic Inclusion Body Myositis. Encyclopedia. Available at: https://encyclopedia.pub/entry/56144. Accessed July 24, 2026.
Guglielmi, Valeria, Marta Cheli, Paola Tonin, Gaetano Vattemi. "Sporadic Inclusion Body Myositis" Encyclopedia, https://encyclopedia.pub/entry/56144 (accessed July 24, 2026).
Guglielmi, V., Cheli, M., Tonin, P., & Vattemi, G. (2024, March 12). Sporadic Inclusion Body Myositis. In Encyclopedia. https://encyclopedia.pub/entry/56144
Guglielmi, Valeria, et al. "Sporadic Inclusion Body Myositis." Encyclopedia. Web. 12 March, 2024.
Copy Citation
Sporadic inclusion body myositis (sIBM) is the most common muscle disease of older people and is clinically characterized by slowly progressive asymmetrical muscle weakness, predominantly affecting the quadriceps, deep finger flexors, and foot extensors.
sporadic inclusion body myositis (sIBM)
inflammation
aging
inflammaging
1. Introduction
Sporadic inclusion body myositis (sIBM) is a chronic progressive muscle disease that primarily affects people 50 years and older [1][2]. It is the most common acquired myopathy in the elderly, with a prevalence ranging between 5 and 180 per million, depending on the geographical area [3][4][5][6]. sIBM is more common in males than females (2:1) and is associated with higher morbidity and mortality [5][6]. The disease presents with muscle weakness mainly affecting the quadriceps and finger flexors [6]. At present, there are no disease-modifying therapies for this progressive disease that eventually leads to severe disability [7]. Despite common clinical characteristics, the phenotype can be variable, and the diagnosis relies on the combination of clinical evaluation, laboratory tests, and pathologic changes in muscle biopsy [6]. Histological hallmarks include both degenerative features such as protein aggregation in myofibers and autoimmune aspects such as endomysial infiltration by T cells [8][9].
sIBM is classified among the idiopathic inflammatory myopathies (IIM) along with dermatomyositis (DM), polymyositis (PM), and immune-mediated necrotizing myopathy (IMNM), but the lack of response to immunosuppressive treatment by sIBM patients have raised questions about the relevance of immune processes in disease pathogenesis [8][10][11].
2. Diagnosis of sIBM
2.1. Clinical Aspects
sIBM is clinically characterized by slowly progressive asymmetrical muscle weakness, predominantly affecting the quadriceps, deep finger flexors, and foot extensors [1][8]. Pharyngeal muscles often become impaired, resulting in dysphagia [12]. Head drop and camptocormia may occur, and facial muscles are occasionally affected [13][14]. Late stages of the disease are characterized by weakness and atrophy of distal and proximal muscles and possible impairment of neck flexors and extensors [15]. Sensory function is normal, and deep tendon reflexes are preserved, but when atrophy of major muscles occurs, the latter can be abolished [16]. Disease progression is usually slow. Variability in the clinical presentation and a lack of specific features/muscle involvement in some patients represent challenges for a prompt and correct diagnosis, making laboratory tests and histological analysis of muscle biopsy fundamental tools in the diagnostic workup [7].
2.2. Laboratory Studies
CK levels are frequently normal or only mildly elevated, usually less than 10 times the upper normal values [9][17].
Autoantibodies are considered a useful tool in the diagnosis of IIM. Based on their specificity, autoantibodies in IIM are traditionally classified as myositis-specific (MSA) or myositis-associated antibodies (MAA), with the latter also occurring in other systemic autoimmune rheumatic diseases [18]. In 2011, Salajegheh et al. showed that autoantibodies targeting a ~44 kDa human muscle protein (referred to as Mup44) occur in the serum of 52% of patients with sIBM (n = 25) with 100% specificity for the disease [19]. In 2013, two independent studies identified Mup44 as cytosolic 5′-nucleotidase 1A (cN1A), which is an enzyme highly expressed in skeletal muscle that catalyzes the dephosphorylation of adenosine monophosphate into adenosine and phosphate [20][21]. Even if absent or extremely rare in healthy controls, antibodies against cN1A have also been detected in primary Sjögren’s syndrome (pSS) and systemic lupus erythematosus (SLE), making them not specific for sIBM [22][23]. Nevertheless, anti-cN1A antibodies are considered a valuable diagnostic biomarker for sIBM because they occur in 33 to 76% of patients with sIBM and in less than 5% of patients with PM, DM, or non-autoimmune neuromuscular diseases [23]. It has been reported that the specificity of anti-cN1A to distinguish sIBM from other IIM, neuromuscular diseases, and autoimmune disorders ranges between 87 and 100% and between 74.6 and 92% to discriminate sIBM from other types of IIM [24]. Contradictory observations have been made regarding the association of anti-cN1A antibodies with a more severe phenotype and increased mortality risk independent of age, gender, comorbidities, and the presence of dysphagia [20][23][24][25][26][27][28][29][30][31]. Even though there is no cure for sIBM, antibodies against cN1A are helpful in the diagnostic workup to reduce misdiagnosis with PM and consequent administration of steroids that worsen sIBM progression after discontinuation [10][15].
2.3. Magnetic Resonance Imaging (MRI)
In recent years, muscle MRI has emerged as an important tool in the diagnosis of muscle disorders due to its ability to highlight affected muscle by revealing atrophy, fat infiltration and signs of inflammation [32]. In sIBM, MRI changes have been reported in both upper and lower limbs, are more severe in distal segments, and usually have an asymmetric pattern [32][33][34][35]. Fat infiltration is commonly seen in MRI scans [33][35][36][37]. It has been observed a distinctive MRI pattern that can discriminate between sIBM and other myopathies, namely the relative sparing of the rectus femoris, hamstrings, and adductor muscles and the extent and asymmetry of fatty infiltration, mostly seen in the adductor magnus [38]. Furthermore, flexor digitorum profundus seems to be involved in sIBM patients on imaging even before its weakness becomes clinically detectable [38].
2.4. Pathological Characteristics
sIBM muscle biopsy shows both degenerative and inflammatory features (Figure 1). Histopathological changes detectable by light microscopy include the following.
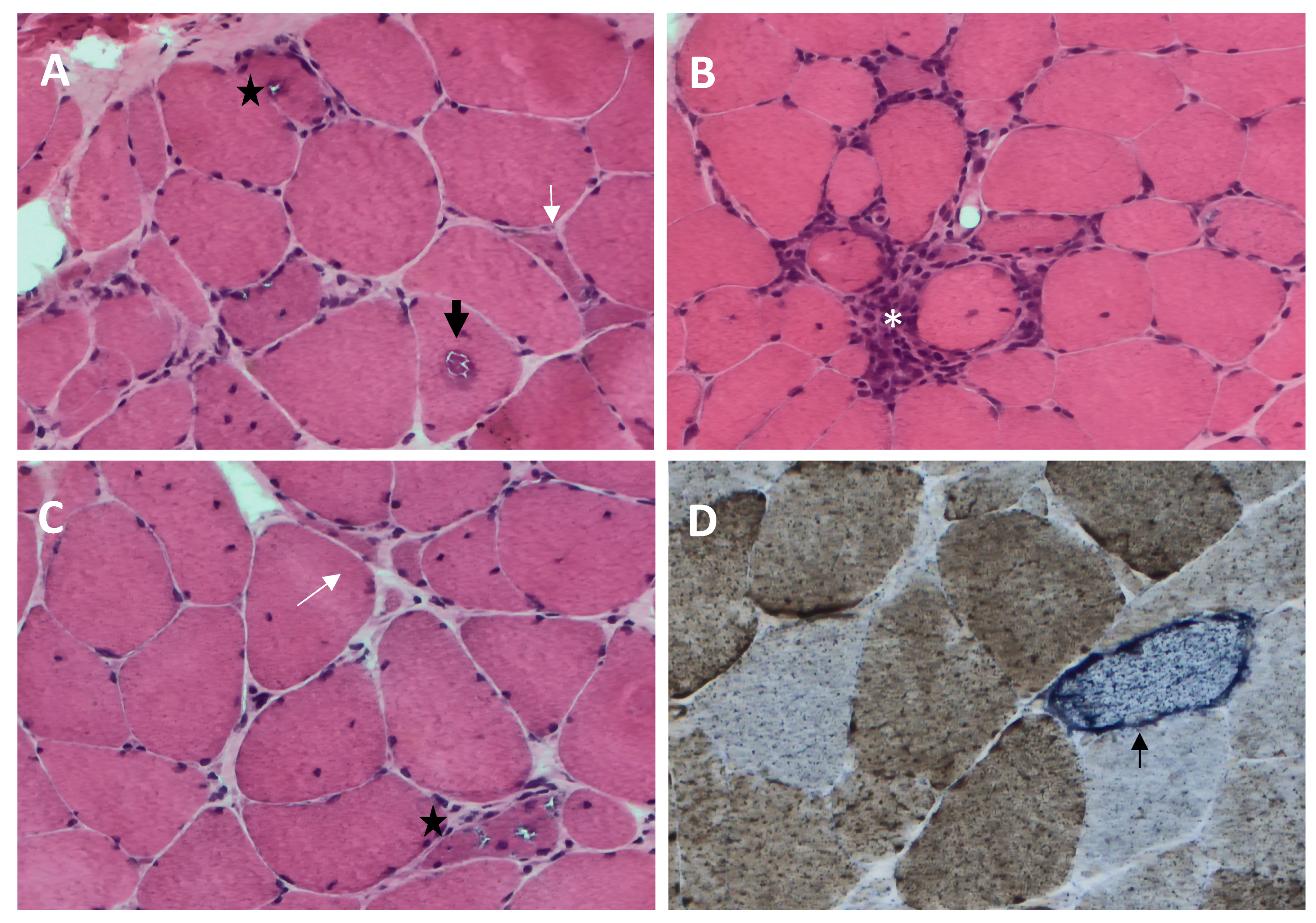
Figure 1. Histopathological features of sporadic inclusion body myositis (sIBM) muscle biopsy ((A–C), hematoxylin and eosin; (D), cytochrome c oxidase (COX)-succinate dehydrogenase): endomysial inflammatory infiltrate that invades nonnecrotic muscle fibers (white asterisk, (B)), rimmed vacuoles within muscle fibers (black star, (A,C)), eosinophilic cytoplasmic inclusion (black arrow, (A)), ragged red fiber and COX-negative fibers (thin black arrow, (D)), and angulated muscle fibers (white arrow, (A,C)).
- (1)
-
Rimmed vacuoles, namely irregular vacuoles of variable size and shape, bordered by basophilic granular deposits that occur in nonnecrotic muscle fibers. Rimmed vacuoles can be a rather rare finding and are visible in 0.4–6.4% of the fibers [9];
- (2)
-
Eosinophilic cytoplasmic inclusions visible at hematoxylin and eosin (H&E) and modified Gomori trichrome staining [17];
- (3)
-
Cytoplasmic accumulation of aggregated/misfolded proteins, referred to as amyloid deposits or protein inclusions, which occur in 60 to 80% of the sIBM vacuolated muscle fibers, usually in non-vacuolated areas of the fiber [1][17]. These β-pleated-sheet amyloid inclusions are detectable by fluorescence-enhanced Congo red staining. Several proteins have been found within these aggregates, including amyloid-β precursor protein (AβPP), amyloid-β (Aβ), phosphorylated-tau (p-tau), and ubiquitin, to name a few [8]. Notably, TDP-43 immunopositivity has been reported in over 60% of sIBM patients and is considered a hallmark of sIBM pathology [39][40];
- (4)
-
Endomysial lymphocytic infiltrates consisting predominantly of macrophages and CD8+T cells that invade nonnecrotic muscle fibers expressing MHC class I on the sarcolemma [17];
- (5)
-
Mitochondrial abnormalities consisting of the abnormal proliferation of mitochondrial leading to ragged red fibers (RRFs, muscle fibers containing excessive mitochondrial proliferation) and the impairment of mitochondrial function, as shown by increased cytochrome c oxidase (COX)-negative fibers [41][42];
- (6)
-
Angulated muscle fibers of small caliber suggesting a neurogenic process [43].
Ultrastructural analysis reveals characteristic degenerative changes such as the following:
- (1)
- (2)
-
Cytoplasmic 6–10 nm amyloid-like filaments containing Aβ, deposits of flocculomembranous and amorphous material [1].
2.5. Correlation of Pathological Features with Clinical Progression and Laboratory Findings
Overall, only a few studies have attempted to correlate specific clinical and/or laboratory findings with muscle histopathological changes. A recent retrospective study on 50 sIBM patients documented a strong and statistically significant positive correlation of endomysial inflammation with the severity of dysphagia and, conversely, a negative correlation with the modified Rankin scale (mRS) whereas the degenerative features such as rimmed vacuoles and congophilic inclusions showed no correlation with any of the clinical measures including muscle strength sum score, quadriceps strength, mRS and severity of dysphagia [44]. Varying and conflicting observations have been made regarding the association of anti-cN1A antibodies with pathological features, thus anti-cN1A antibodies correlated with fewer rimmed vacuoles [45], more cytochrome oxidase negative myofibers [25][46], a higher number of regenerating fibers [44], or auto-aggressive inflammation [47]. CK levels correlated with a broad spectrum of histological findings, including muscle fiber necrosis, regeneration, endomysial and perimysial inflammation, and increased endomysial connective tissue [44].
3. Inflammation in sIBM Pathogenesis
The involvement of the immune system in sIBM is supported by the presence of endomysial immune cell infiltration and circulating autoantibodies. Although the role of inflammation in the pathogenesis of sIBM has been under discussion for a long time, many studies have provided data to support the involvement of both innate and adaptive immunity in the disease.
3.1. T Cells
Infiltrating immune cells are mostly represented by cytotoxic CD8+T cells that surround non-necrotic muscle fibers and are often localized in proximity to MHCI-expressing myofibers [17]. It has been estimated that CD8+ T cells are about fivefold more abundant than CD4+ T cells in sIBM muscle [48]. Several studies have shown that CD8+ T cells have restricted expression of T cell receptor (TCR) Vα/Vβ genes in the blood and muscle of sIBM patients and are clonally expanded, suggesting that muscle-specific antigen drives the inflammatory response in the disease [49][50][51][52][53][54][55][56][57]. Interestingly, the expression of Vβ genes was found to be different in muscle-infiltrating lymphocytes and peripheral blood, indicating that T cells expand in situ in response to antigens presented by MHCI-expressing muscle cells, or they are selectively recruited to the muscle [52][53]. Identical T cell clones have been found in different muscles and persist for years, indicating that they continuously react to muscle antigens, perpetuating the inflammatory response over time [51][58].
T cell activation requires antigen presentation via MHC and the interaction between CD28 on T cells with costimulatory molecules on antigen-presenting cells (APCs). Expression of the costimulatory molecules BB-1, ICOS, and B7-H3 by muscle fibers has been demonstrated in sIBM and suggests that myofibers may present the antigen and activate T cells expressing the appropriate TCR [59][60][61][62].
Regarding phenotypic and functional features, infiltrating CD8+ T cells express markers of highly differentiated effector cells, have proinflammatory and cytotoxic features, and have minimal or no proliferative ability [54][63][64][65][66]. Specifically, expression of the cytolytic molecules perforin, granulysin, and granzyme B, and natural killer (NK) markers CD16 or CD56, have been detected in CD8+T cells invading skeletal muscle, indicating they share functional and phenotypic features of NK cells [61][67][68][69]. The occurrence of Th1 immune response has been supported by increased expression levels of the cytokines and chemokines CXCL-9, CXCL-10, IL-12, CCL-2, and IL-1RA and by a higher number of IFN-γ expressing CD8+CD28− T cells in the blood of patients with sIBM [65]. These findings indicate that cytotoxic T cells respond to yet unknown muscle antigen/s and contribute to the myofiber damage that occurs in sIBM [9].
On the other hand, several observations show that, in sIBM, T cells are highly differentiated and have reduced effector functions. In this regard, it has been found that peripheral blood and muscle infiltrating T cells are effector memory T (TEM) cells and terminally differentiated effector memory T (TEMRA), as indicated by the lack of expression of CD28 and by the acquisition of CD224 and CD57 expression [9][65][70]. Loss of CD28 and gain of CD57 in T cells are known to be associated with decreased proliferative capacity, terminal differentiation, senescence, and clonal exhaustion, a state occurring with chronic antigen stimulation [71][72]. Exhausted T-cells have decreased ability to respond to cytokines, have lost their effector functions, and have upregulated inhibitory receptors (IRs) such as programmed cell death protein1 (PD-1), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin domain and mucin domain-3 (TIM-3), and T-cell immune receptor with Ig and ITIM domains (TIGIT) [70][73][74]. The main function of IRs is to negatively regulate the activation and the effector functions of T cells and are responsible for fine-tuning T cell activity [74]. IRs have a key role in controlling immune responses and establishing T cell tolerance [74]. The expression of PD-1 has been reported on T cells infiltrating sIBM muscle [75]. PD1 ligands PD-L1 and PD-L2 have been detected on infiltrating macrophages and on myofibers, respectively, and possibly form immunologic synapses with PD1 expressed by lymphocytes [75]. In support of an exhausted phenotype of T cells in sIBM, the mRNA levels of the exhaustion markers PD1, EOMES, TBX21, LAG3, CD244, TIM3, and KLRG1 were found to be increased in sIBM muscle [75]. Taken together, these data indicate that, in sIBM, persistent exposure to antigens drives muscle infiltrating T cells toward a state of exhaustion characterized by reduced or absent effector functions and proliferation ability. These observations suggest that the T cell-mediated cytotoxic damage of muscle fibers likely plays a role in the early stages of the disease before T cell exhaustion occurs.
Regulatory T cells (Tregs) represent a subpopulation of CD4+T cells which have the main function to suppress immune responses [76]. Their immune suppressive activity is crucial for keeping immune reactions against invading pathogens under control and maintaining immune tolerance toward self-antigens [76]. The decreased number and function of Tregs have been associated with loss of tolerance and autoimmune diseases [77]. Studies have demonstrated that Tregs are able to control local immune responses during regeneration after skeletal muscle injury [78][79]. Although Tregs suppression activity in sIBM was found to be normal, a reduction in their number was observed in the blood and skeletal muscle of sIBM patients and proposed as a factor contributing to the autoimmune response to skeletal muscle in this disease [65].
3.2. Plasma Cells and Antibody-Mediated Immune Response
Several studies have provided evidence for a muscle antigen-driven B- and plasma cell-mediated immune response and humoral immunity in sIBM. Differentiated CD138+ plasma cells, but not CD19+ or CD20+ B cells, and increased expression of immunoglobulin gene transcripts were detected in the skeletal muscle of patients with sIBM [80]. Analysis of the variable region of Ig H chain gene transcript obtained from sIBM muscles demonstrated that B cells and their descendent plasma cells undergo oligoclonal expansion, isotype switching, and accumulate somatic mutations [81]. These processes usually occur in secondary lymphoid organs where B cells migrate after exposure to antigens to maturate and differentiate into antibody-producing plasma cells [81]. Interestingly, it has been observed that in sIBM muscles, inflammatory infiltrates of T cells, B cells, plasma cells, and myeloid dendritic cells assemble in lymphoid structures that could support the in situ maturation of B cells into antibody-secreting plasma cells [82]. Furthermore, increased serum levels of B-cell activating factor (BAFF), a cytokine involved in the survival, maturation, and differentiation of B cells, were detected in the serum of patients with sIBM [83]. These findings indicate that local maturation of B cells to antibody-producing plasma cells occurs in the muscle of sIBM patients in response to muscle antigen/s [81]. In agreement with these observations, a recent study revealed the sIBM muscle B cell receptor (BCR) repertoire and showed that it has distinct features from those of other IIMs, indicating that it may originate from exposure to disease-specific antigens [84].
The presence of a humoral immune response in sIBM was further supported by the detection of antibodies against cN1A, as described previously in this manuscript. The pathogenic role of the anti-cN1A was supported by the finding that mice immunized with anti-cN1A IgG isolated from patients show increased sarcoplasmic aggregation of p62 and LC3, degenerative features of sIBM muscle [30].
Overall, these findings support that in sIBM, B cells become activated and mature into plasma cells that produce antibodies directed against a muscle antigen.
3.3. Macrophages and Dendritic Cells
Macrophages and myeloid dendritic cells (DCs) have also been detected in sIBM muscle [48][85]. Proteomic studies have shown that CD74, CD163, and STAT1 are increased in sIBM muscle, and immunohistochemical analysis revealed these molecules are enriched in macrophages [86]. Specifically, CD74 was found to be expressed in CD68+ macrophages and the sarcolemma. CD163 was detected in endomysial macrophages, and STAT1 was found in macrophages that displayed phagocytic activity [86]. The interferon-induced protein ISG15 and IRF8 were found to be strongly expressed on MHCII+ macrophages, indicating that these cells are in an activated state [86].
DCs are also infiltrating the sIBM muscle. A study of the two populations of DCs, namely myeloid DC (mDCs) and plasmacytoid DC (pDCs), revealed that mDCs surround and invade healthy myofibers [87][88]. The close colocalization of mDCs with T cells has been interpreted as a possible role of mDCs in antigen presentation to T cells [87][88].
These studies show that, despite the expression of MHC on muscle fibers of sIBM patients and their possible contribution to antigen presentation, professional antigen-presenting cells (APCs) also play a role in the pathogenesis of sIBM [89][90][91]. However, further efforts are needed to better characterize the phenotype and function of macrophages and dendritic cells in sIBM.
References
- Engel, W.K.; Askanas, V. Inclusion-body myositis: Clinical, diagnostic, and pathologic aspects. Neurology 2006, 66, S20–S29.
- Carpenter, S.; Karpati, G.; Heller, I.; Eisen, A. Inclusion body myositis: A distinct variety of idiopathic inflammatory myopathy. Neurology 1978, 28, 8–17.
- Price, M.A.; Barghout, V.; Benveniste, O.; Christopher-Stine, L.; Corbett, A.; de Visser, M.; Hilton-Jones, D.; Kissel, J.T.; Lloyd, T.E.; Lundberg, I.E.; et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: Survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. J. Neuromuscul. Dis. 2016, 3, 67–75.
- Needham, M.; Corbett, A.; Day, T.; Christiansen, F.; Fabian, V.; Mastaglia, F.L. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J. Clin. Neurosci. 2008, 15, 1350–1353.
- Callan, A.; Capkun, G.; Vasanthaprasad, V.; Freitas, R.; Needham, M. A Systematic Review and Meta-Analysis of Prevalence Studies of Sporadic Inclusion Body Myositis. J. Neuromuscul. Dis. 2017, 4, 127–137.
- Nagy, S.; Khan, A.; Machado, P.M.; Houlden, H. Inclusion body myositis: From genetics to clinical trials. J. Neurol. 2023, 270, 1787–1797.
- Skolka, M.P.; Naddaf, E. Exploring challenges in the management and treatment of inclusion body myositis. Curr. Opin. Rheumatol. 2023, 35, 404–413.
- Askanas, V.; Engel, W.K.; Nogalska, A. Sporadic inclusion-body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim. Biophys. Acta 2015, 1852, 633–643.
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272.
- Benveniste, O.; Guiguet, M.; Freebody, J.; Dubourg, O.; Squier, W.; Maisonobe, T.; Stojkovic, T.; Leite, M.I.; Allenbach, Y.; Herson, S.; et al. Long-term observational study of sporadic inclusion body myositis. Brain 2011, 134, 3176–3184.
- Danon, M.J.; Reyes, M.G.; Perurena, O.H.; Masdeu, J.C.; Manaligod, J.R. Inclusion body myositis. A corticosteroid-resistant idiopathic inflammatory myopathy. Arch. Neurol. 1982, 39, 760–764.
- Oh, T.H.; Brumfield, K.A.; Hoskin, T.L.; Kasperbauer, J.L.; Basford, J.R. Dysphagia in inclusion body myositis: Clinical features, management, and clinical outcome. Am. J. Phys. Med. Rehabil. 2008, 87, 883–889.
- Goodman, B.P.; Liewluck, T.; Crum, B.A.; Spinner, R.J. Camptocormia due to inclusion body myositis. J. Clin. Neuromuscul. Dis. 2012, 14, 78–81.
- Salam, S.; Morrow, J.M.; Howard, R.; Miller, J.A.L.; Quinlivan, R.M.; Machado, P.M. Two emerging phenotypes of atypical inclusion body myositis: Illustrative cases. Clin. Exp. Rheumatol. 2023, 41, 340–347.
- Naddaf, E. Inclusion body myositis: Update on the diagnostic and therapeutic landscape. Front. Neurol. 2022, 13, 1020113.
- Beyenburg, S.; Zierz, S.; Jerusalem, F. Inclusion body myositis: Clinical and histopathological features of 36 patients. Clin. Investig. 1993, 71, 351–361.
- Dimachkie, M.M. Idiopathic inflammatory myopathies. J. Neuroimmunol. 2011, 231, 32–42.
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin. Rev. Allergy Immunol. 2017, 52, 1–19.
- Salajegheh, M.; Lam, T.; Greenberg, S.A. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PLoS ONE 2011, 6, e20266.
- Larman, H.B.; Salajegheh, M.; Nazareno, R.; Lam, T.; Sauld, J.; Steen, H.; Kong, S.W.; Pinkus, J.L.; Amato, A.A.; Elledge, S.J.; et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann. Neurol. 2013, 73, 408–418.
- Pluk, H.; van Hoeve, B.J.; van Dooren, S.H.; Stammen-Vogelzangs, J.; van der Heijden, A.; Schelhaas, H.J.; Verbeek, M.M.; Badrising, U.A.; Arnardottir, S.; Gheorghe, K.; et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann. Neurol. 2013, 73, 397–407.
- Rietveld, A.; van den Hoogen, L.L.; Bizzaro, N.; Blokland, S.L.M.; Dähnrich, C.; Gottenberg, J.E.; Houen, G.; Johannsen, N.; Mandl, T.; Meyer, A.; et al. Autoantibodies to Cytosolic 5′-Nucleotidase 1A in Primary Sjögren’s Syndrome and Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1200.
- Herbert, M.K.; Stammen-Vogelzangs, J.; Verbeek, M.M.; Rietveld, A.; Lundberg, I.E.; Chinoy, H.; Lamb, J.A.; Cooper, R.G.; Roberts, M.; Badrising, U.A.; et al. Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann. Rheum. Dis. 2016, 75, 696–701.
- Diederichsen, L.P.; Iversen, L.V.; Nielsen, C.T.; Jacobsen, S.; Hermansen, M.L.; Witting, N.; Cortes, R.; Korsholm, S.S.; Krogager, M.E.; Friis, T. Myositis-related autoantibody profile and clinical characteristics stratified by anti-cytosolic 5′-nucleotidase 1A status in connective tissue diseases. Muscle Nerve 2023, 68, 73–80.
- Lilleker, J.B.; Rietveld, A.; Pye, S.R.; Mariampillai, K.; Benveniste, O.; Peeters, M.T.; Miller, J.A.; Hanna, M.G.; Machado, P.M.; Parton, M.J.; et al. Cytosolic 5′-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann. Rheum. Dis. 2017, 76, 862–868.
- Goyal, N.A.; Cash, T.M.; Alam, U.; Enam, S.; Tierney, P.; Araujo, N.; Mozaffar, F.H.; Pestronk, A.; Mozaffar, T. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J. Neurol. Neurosurg. Psychiatry 2016, 87, 373–378.
- Lucchini, M.; Maggi, L.; Pegoraro, E.; Filosto, M.; Rodolico, C.; Antonini, G.; Garibaldi, M.; Valentino, M.L.; Siciliano, G.; Tasca, G.; et al. Anti-cN1A Antibodies Are Associated with More Severe Dysphagia in Sporadic Inclusion Body Myositis. Cells 2021, 10, 1146.
- Felice, K.J.; Whitaker, C.H.; Wu, Q.; Larose, D.T.; Shen, G.; Metzger, A.L.; Barton, R.W. Sensitivity and clinical utility of the anti-cytosolic 5′-nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: Report of 40 patients from a single neuromuscular center. Neuromuscul. Disord. 2018, 28, 660–664.
- Oyama, M.; Ohnuki, Y.; Inoue, M.; Uruha, A.; Yamashita, S.; Yutani, S.; Tanboon, J.; Nakahara, J.; Suzuki, S.; Shiina, T.; et al. HLA-DRB1 allele and autoantibody profiles in Japanese patients with inclusion body myositis. PLoS ONE 2020, 15, e0237890.
- Tawara, N.; Yamashita, S.; Zhang, X.; Korogi, M.; Zhang, Z.; Doki, T.; Matsuo, Y.; Nakane, S.; Maeda, Y.; Sugie, K.; et al. Pathomechanisms of anti-cytosolic 5′-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann. Neurol. 2017, 81, 512–525.
- Greenberg, S.A. Cytoplasmic 5′-nucleotidase autoantibodies in inclusion body myositis: Isotypes and diagnostic utility. Muscle Nerve 2014, 50, 488–492.
- Zubair, A.S.; Salam, S.; Dimachkie, M.M.; Machado, P.M.; Roy, B. Imaging biomarkers in the idiopathic inflammatory myopathies. Front. Neurol. 2023, 14, 1146015.
- Dion, E.; Cherin, P.; Payan, C.; Fournet, J.C.; Papo, T.; Maisonobe, T.; Auberton, E.; Chosidow, O.; Godeau, P.; Piette, J.C.; et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J. Rheumatol. 2002, 29, 1897–1906.
- Phillips, B.A.; Cala, L.A.; Thickbroom, G.W.; Melsom, A.; Zilko, P.J.; Mastaglia, F.L. Patterns of muscle involvement in inclusion body myositis: Clinical and magnetic resonance imaging study. Muscle Nerve 2001, 24, 1526–1534.
- Guimaraes, J.B.; Zanoteli, E.; Link, T.M.; de Camargo, L.V.; Facchetti, L.; Nardo, L.; Fernandes, A. Sporadic Inclusion Body Myositis: MRI Findings and Correlation With Clinical and Functional Parameters. Am. J. Roentgenol. 2017, 209, 1340–1347.
- Ansari, B.; Salort-Campana, E.; Ogier, A.; Le Troter Ph, D.A.; De Sainte Marie, B.; Guye, M.; Delmont, E.; Grapperon, A.M.; Verschueren, A.; Bendahan, D.; et al. Quantitative muscle MRI study of patients with sporadic inclusion body myositis. Muscle Nerve 2020, 61, 496–503.
- Tasca, G.; Monforte, M.; De Fino, C.; Kley, R.A.; Ricci, E.; Mirabella, M. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve 2015, 52, 956–962.
- Cox, F.M.; Reijnierse, M.; van Rijswijk, C.S.; Wintzen, A.R.; Verschuuren, J.J.; Badrising, U.A. Magnetic resonance imaging of skeletal muscles in sporadic inclusion body myositis. Rheumatology 2011, 50, 1153–1161.
- Hiniker, A.; Daniels, B.H.; Lee, H.S.; Margeta, M. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol. Commun. 2013, 1, 29.
- Askanas, V.; Engel, W.K.; Nogalska, A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J. Neuropathol. Exp. Neurol. 2012, 71, 680–693.
- Lindgren, U.; Roos, S.; Hedberg Oldfors, C.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 2015, 25, 281–288.
- Oldfors, A.; Moslemi, A.R.; Jonasson, L.; Ohlsson, M.; Kollberg, G.; Lindberg, C. Mitochondrial abnormalities in inclusion-body myositis. Neurology 2006, 66, S49–S55.
- Vattemi, G.; Mirabella, M.; Guglielmi, V.; Lucchini, M.; Tomelleri, G.; Ghirardello, A.; Doria, A. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Autoimmun. Highlights 2014, 5, 77–85.
- Pinto, M.V.; Laughlin, R.S.; Klein, C.J.; Mandrekar, J.; Naddaf, E. Inclusion body myositis: Correlation of clinical outcomes with histopathology, electromyography and laboratory findings. Rheumatology 2022, 61, 2504–2511.
- Lloyd, T.E.; Christopher-Stine, L.; Pinal-Fernandez, I.; Tiniakou, E.; Petri, M.; Baer, A.; Danoff, S.K.; Pak, K.; Casciola-Rosen, L.A.; Mammen, A.L. Cytosolic 5′-Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis Care Res. 2016, 68, 66–71.
- Ikenaga, C.; Findlay, A.R.; Goyal, N.A.; Robinson, S.; Cauchi, J.; Hussain, Y.; Wang, L.H.; Kershen, J.C.; Beson, B.A.; Wallendorf, M.; et al. Clinical utility of anti-cytosolic 5′-nucleotidase 1A antibody in idiopathic inflammatory myopathies. Ann. Clin. Transl. Neurol. 2021, 8, 571–578.
- Paul, P.; Liewluck, T.; Ernste, F.C.; Mandrekar, J.; Milone, M. Anti-cN1A antibodies do not correlate with specific clinical, electromyographic, or pathological findings in sporadic inclusion body myositis. Muscle Nerve 2021, 63, 490–496.
- Engel, A.G.; Arahata, K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann. Neurol. 1984, 16, 209–215.
- Salajegheh, M.; Rakocevic, G.; Raju, R.; Shatunov, A.; Goldfarb, L.G.; Dalakas, M.C. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 2007, 69, 1672–1679.
- Hofbauer, M.; Wiesener, S.; Babbe, H.; Roers, A.; Wekerle, H.; Dornmair, K.; Hohlfeld, R.; Goebels, N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 4090–4095.
- Müntzing, K.; Lindberg, C.; Moslemi, A.R.; Oldfors, A. Inclusion body myositis: Clonal expansions of muscle-infiltrating T cells persist over time. Scand. J. Immunol. 2003, 58, 195–200.
- Fyhr, I.M.; Moslemi, A.R.; Tarkowski, A.; Lindberg, C.; Oldfors, A. Limited T-cell receptor V gene usage in inclusion body myositis. Scand. J. Immunol. 1996, 43, 109–114.
- Fyhr, I.M.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. T cell receptor beta-chain repertoire in inclusion body myositis. J. Neuroimmunol. 1998, 91, 129–134.
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur. J. Immunol. 1994, 24, 2659–2663.
- O’Hanlon, T.P.; Dalakas, M.C.; Plotz, P.H.; Miller, F.W. The alpha beta T-cell receptor repertoire in inclusion body myositis: Diverse patterns of gene expression by muscle-infiltrating lymphocytes. J. Autoimmun. 1994, 7, 321–333.
- Fyhr, I.M.; Moslemi, A.R.; Mosavi, A.A.; Lindberg, C.; Tarkowski, A.; Oldfors, A. Oligoclonal expansion of muscle infiltrating T cells in inclusion body myositis. J. Neuroimmunol. 1997, 79, 185–189.
- Bender, A.; Behrens, L.; Engel, A.G.; Hohlfeld, R. T-cell heterogeneity in muscle lesions of inclusion body myositis. J. Neuroimmunol. 1998, 84, 86–91.
- Amemiya, K.; Granger, R.P.; Dalakas, M.C. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 2000, 123 Pt 10, 2030–2039.
- Murata, K.; Dalakas, M.C. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am. J. Pathol. 1999, 155, 453–460.
- Behrens, L.; Kerschensteiner, M.; Misgeld, T.; Goebels, N.; Wekerle, H.; Hohlfeld, R. Human muscle cells express a functional costimulatory molecule distinct from B7.1 (CD80) and B7.2 (CD86) in vitro and in inflammatory lesions. J. Immunol. 1998, 161, 5943–5951.
- Schmidt, J.; Rakocevic, G.; Raju, R.; Dalakas, M.C. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: Significance for CD8+ T cell cytotoxicity. Brain 2004, 127, 1182–1190.
- Waschbisch, A.; Wintterle, S.; Lochmüller, H.; Walter, M.C.; Wischhusen, J.; Kieseier, B.C.; Wiendl, H. Human muscle cells express the costimulatory molecule B7-H3, which modulates muscle-immune interactions. Arthritis Rheumatol. 2008, 58, 3600–3608.
- Pandya, J.M.; Fasth, A.E.; Zong, M.; Arnardottir, S.; Dani, L.; Lindroos, E.; Malmström, V.; Lundberg, I.E. Expanded T cell receptor Vβ-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheumatol. 2010, 62, 3457–3466.
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Local T-cell proliferation and differentiation in inflammatory myopathies. Scand. J. Immunol. 1995, 41, 421–426.
- Allenbach, Y.; Chaara, W.; Rosenzwajg, M.; Six, A.; Prevel, N.; Mingozzi, F.; Wanschitz, J.; Musset, L.; Charuel, J.L.; Eymard, B.; et al. Th1 response and systemic treg deficiency in inclusion body myositis. PLoS ONE 2014, 9, e88788.
- Greenberg, S.A.; Pinkus, J.L.; Kong, S.W.; Baecher-Allan, C.; Amato, A.A.; Dorfman, D.M. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 2019, 142, 2590–2604.
- Ikezoe, K.; Ohshima, S.; Osoegawa, M.; Tanaka, M.; Ogawa, K.; Nagata, K.; Kira, J.I. Expression of granulysin in polymyositis and inclusion-body myositis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1187–1190.
- Orimo, S.; Koga, R.; Goto, K.; Nakamura, K.; Arai, M.; Tamaki, M.; Sugita, H.; Nonaka, I.; Arahata, K. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul. Disord. 1994, 4, 219–226.
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Invest. 1996, 97, 2905–2910.
- Matsubara, S.; Suzuki, S.; Komori, T. Immunohistochemical Phenotype of T Cells Invading Muscle in Inclusion Body Myositis. J. Neuropathol. Exp. Neurol. 2022, 81, 825–835.
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32.
- Dzangué-Tchoupou, G.; Mariampillai, K.; Bolko, L.; Amelin, D.; Mauhin, W.; Corneau, A.; Blanc, C.; Allenbach, Y.; Benveniste, O. CD8+(T-bet+) cells as a predominant biomarker for inclusion body myositis. Autoimmun. Rev. 2019, 18, 325–333.
- Gao, Z.; Feng, Y.; Xu, J.; Liang, J. T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy. Front. Immunol. 2022, 13, 977394.
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499.
- Knauss, S.; Preusse, C.; Allenbach, Y.; Leonard-Louis, S.; Touat, M.; Fischer, N.; Radbruch, H.; Mothes, R.; Matyash, V.; Böhmerle, W.; et al. PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol. Neuroimmunol. Neuroinflammation 2019, 6, e558.
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015.
- Valentini, N.; Requejo Cier, C.J.; Lamarche, C. Regulatory T-cell dysfunction and its implication for cell therapy. Clin. Exp. Immunol. 2023, 213, 40–49.
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295.
- Schiaffino, S.; Pereira, M.G.; Ciciliot, S.; Rovere-Querini, P. Regulatory T cells and skeletal muscle regeneration. FEBS J. 2017, 284, 517–524.
- Greenberg, S.A.; Bradshaw, E.M.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Due, B.; Bregoli, L.; O’Connor, K.C.; Amato, A.A. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005, 65, 1782–1787.
- Bradshaw, E.M.; Orihuela, A.; McArdel, S.L.; Salajegheh, M.; Amato, A.A.; Hafler, D.A.; Greenberg, S.A.; O’Connor, K.C. A local antigen-driven humoral response is present in the inflammatory myopathies. J. Immunol. 2007, 178, 547–556.
- Salajegheh, M.; Pinkus, J.L.; Amato, A.A.; Morehouse, C.; Jallal, B.; Yao, Y.; Greenberg, S.A. Permissive environment for B-cell maturation in myositis muscle in the absence of B-cell follicles. Muscle Nerve 2010, 42, 576–583.
- Krystufková, O.; Vallerskog, T.; Helmers, S.B.; Mann, H.; Putová, I.; Belácek, J.; Malmström, V.; Trollmo, C.; Vencovsky, J.; Lundberg, I.E. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann. Rheum. Dis. 2009, 68, 836–843.
- Jiang, R.; Roy, B.; Wu, Q.; Mohanty, S.; Nowak, R.J.; Shaw, A.C.; Kleinstein, S.H.; O’Connor, K.C. The Plasma Cell Infiltrate Populating the Muscle Tissue of Patients with Inclusion Body Myositis Features Distinct B Cell Receptor Repertoire Properties. Immunohorizons 2023, 7, 310–322.
- Benveniste, O.; Stenzel, W.; Hilton-Jones, D.; Sandri, M.; Boyer, O.; van Engelen, B.G. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: The inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015, 129, 611–624.
- Roos, A.; Preusse, C.; Hathazi, D.; Goebel, H.H.; Stenzel, W. Proteomic Profiling Unravels a Key Role of Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Front. Immunol. 2019, 10, 1040.
- Greenberg, S.A.; Pinkus, G.S.; Amato, A.A.; Pinkus, J.L. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve 2007, 35, 17–23.
- de Padilla, C.M.; Reed, A.M. Dendritic cells and the immunopathogenesis of idiopathic inflammatory myopathies. Curr. Opin. Rheumatol. 2008, 20, 669–674.
- Das, L.; Blumbergs, P.C.; Manavis, J.; Limaye, V.S. Major histocompatibility complex class I and II expression in idiopathic inflammatory myopathy. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 539–542.
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am. J. Pathol. 2001, 159, 1263–1273.
- Bartoccioni, E.; Gallucci, S.; Scuderi, F.; Ricci, E.; Servidei, S.; Broccolini, A.; Tonali, P. MHC class I, MHC class II and intercellular adhesion molecule-1 (ICAM-1) expression in inflammatory myopathies. Clin. Exp. Immunol. 1994, 95, 166–172.
More
Information
Subjects:
Clinical Neurology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
750
Revisions:
2 times
(View History)
Update Date:
12 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No