 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Homer Selton Black | -- | 4295 | 2024-03-10 20:46:25 | | | |
| 2 | Jason Zhu | -81 word(s) | 4214 | 2024-03-12 03:37:35 | | |
Video Upload Options
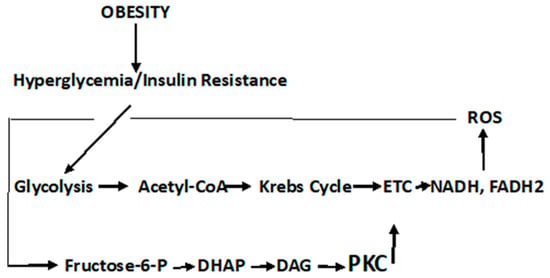
Type 2 diabetes mellitus (T2DM) accounts for one-sixth of deaths globally, whereas cancer is the second leading cause of death in the U.S. T2DM is a known risk factor for many cancers. Reactive oxygen species (ROS)-altered metabolic and signaling pathways link T2DM to cancer. These reprogrammed metabolic and signaling pathways contribute to diabetic complications, impact the redox balance (oxidative stress), and have differential roles in the early and late stages of cancer. A respiratory chain that is highly reduced (as under hyperglycemic conditions) or if reduced cofactors accumulate, ROS are greatly elevated. ROS may cause mutations in mitochondrial DNA (mtDNA) that result in further ROS elevations. The amplification of ROS results in the activation of PKC, an overarching signaling pathway that activates MAPK with a subsequent regulation in several factors that result in pathophysiological manifestations of T2DM and cancer. An upregulation in PKC leads to a deregulation in NF-kß, which regulates the PKB/P13/Akt pathway and orchestrates the cell survival, growth, proliferation, and glucose metabolism manifested in cancer. It also affects Insulin Receptor Substrate (IRS-1), decreasing insulin-stimulated glucose transport and glucose uptake, disrupting subsequent cell signaling pathways contributing to the development of T2DM.
1. Diabetic Metabolism Linking Cancer


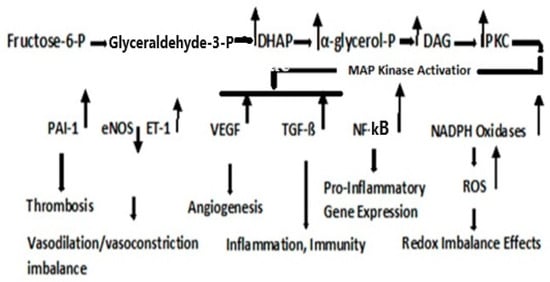
2. Dyslipidemia and Cell Signaling
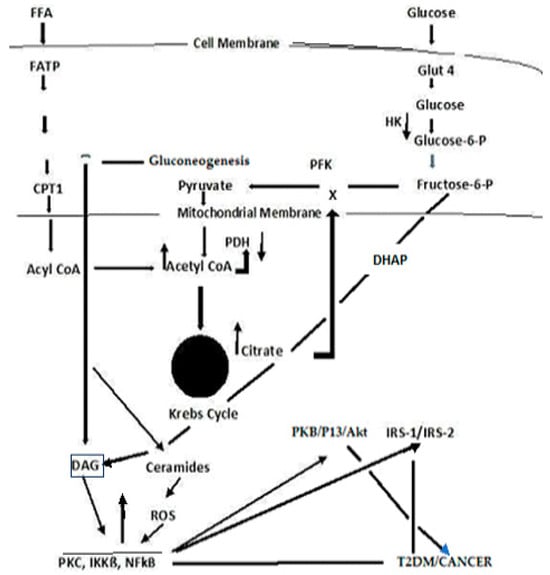
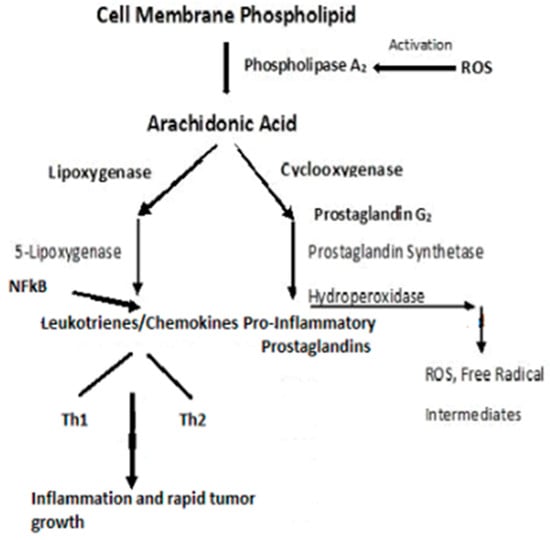
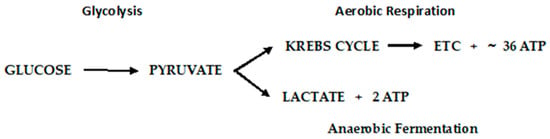
3. Cancer Cell Metabolism, ROS, and Antioxidant Therapy
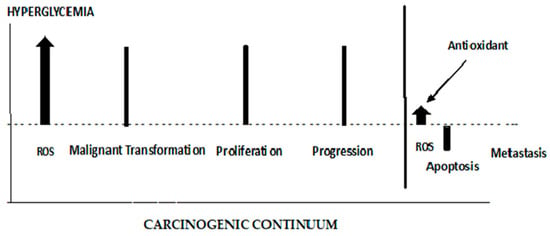
4. ROS’ Role in the Early and Late Stages of Cancer
References
- Black, H.S. A Synopsis of the Associations of Oxidative Stress, ROS, and Antioxidants with Diabetes Mellitus. Antioxidants 2022, 11, 2003.
- Brownlee, M. The Pathology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625.
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13.
- Schleicher, E.D.; Weigert, C. Role of hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. 2000, 58 (Suppl. S77), S13–S18.
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513.
- Turkmen, K.; Karagoz, A.; Kucuk, A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J. Diabetes 2014, 5, 894–900.
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.J. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. 2016, 10, 145–153.
- Szabo, C.; Biser, A.; Benko, R.; Bottinger, E.; Susztak, K. Poly (ADP-Ribose) Polymerase Inhibitors Ameliorate Nephropathy of Type 2 Diabetic Leprdb/db Mice. Diabetes 2006, 55, 3004–3012.
- McGarry, J.D. Banting Lecture 2001: Dysregulation of fatty acid metabolism in the etiology of Type 2 diabetes. Diabetes 2002, 51, 7–18.
- Reaven, G.M. Role of Insulin Resistance in Human Disease (Syndrome X): An Expanded Definition. Annu. Rev. Med. 1993, 44, 121–131.
- Lau, E.S.; Paniagua, S.M.; Liu, E.; Jovani, M.; Li, S.X.; Takvorian, K.; Suthahar, N.; Cheng, S.; Splansky, G.L.; Januzzi, J.L.; et al. Shared risk factors in cardiovascular disease and cancer. J. Am. Coll. Cardiol. 2021, 3, 48–58.
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared risk factors in cardiovascular disease and cancer. Circulation. 2016, 133, 1104–1114.
- Duarte, C.W.; Lindner, V.; Francis, S.A.; Schoormans, D. Visualization of cancer and cardiovascular disease co-occurrence with network methods. JCO Clin. Cancer Inform. 2017, 1, 1–12.
- Katzie, V.A.; Sookthai, D.; Johnson, T.; Kühn, T.; Kaaks, R. Blood lipids and lipoproteins in relation to incidence and mortality risks for CVD and cancer in the prospective epic-Heidelberg cohort. BMC Med. 2017, 15, 218.
- Nomikos, T.; Panagiotakos, D.; Georgousopoulou, E.; Metaxa, V.; Chrysohoou, C.; Skoumas, I.; Antonopoulou, S.; Tousoulis, D.; Stefanadis, C.; Pitsavos, C.; et al. Hierarchical modelling of blood lipids’ profile and 10-year (2002–2012) all-cause mortality and incidence of cardiovascular disease: The Attica study. Lipids Health Dis. 2015, 14, 108.
- Hasbani, N.R.; Ligthart, S.; Brown, M.R.; Heath, A.S.; Bebo, A.; Ashley, K.E.; Boerwinkle, E.; Morrison, A.C.; Folsom, A.R.; Aguilar, D.; et al. American Heart Association’s life’s simple 7: Lifestyle recommendations, polygenic risk, and lifetime risk of coronary heart disease. Circulation 2022, 145, 808–818.
- Mooradian, A.D. Dyslipidemia in type 2 diabetes mellitus. Nat. Clin. Prac. Endocrinol. Metab. 2009, 5, 150–159.
- Korac, B.; Kalezic, A.; Pekovic-Vaughan, V.; Koras, A. Redox changes in obesity, metabolic syndrome, and diabetes. Redox Biol. 2021, 42, 101887.
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789.
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496.
- Delarue, J.; Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148.
- Houten, S.M.; Wanders, R.J.A. General introduction to the biochemistry of mitochondrial fatty acid ß-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477.
- Kaur, S.; Auger, C.; Jeschke, M.G. Adipose tissue metabolic function and dysfunction: Impact of burn injury. Front. Cell Dev. Biol. 2020, 8, 599576.
- De Souza Bastros, A.; Graves, D.T.; de Melo Loureiro, A.P.; Junior, C.R.; Corbi, S.C.T.; Frizzera, F.; Orrico, S.R.P. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Kim, D.; Chung, H.; Lee, J.E.; Kim, J.; Hwang, J.; Chung, Y. Immunologic Aspects of Dyslipidemia: A Critical Regulator of Adaptive Immunity and Immune Disorders. J. Lipid Atheroscler. 2021, 10, 184–201.
- Berger, A. Th1 and Th2 responses: What are they? Brit. Med. J. 2000, 321, 424.
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971.
- Ledford, H. How air pollution causes lung cancer—Without harming DNA. Nature 2023, 616, 419–420.
- Kang, J.-H. Protein kinase C (PKC) isozymes and cancer. New J. Sci. 2014, 2014, 231418.
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.B.; Abba, M.; Kazanietz, M.G. Protein Kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237.
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52.
- Griner, E.; Kazanietz, M. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294.
- Jemal, A.; Torre, L.; Soerjomataram, I.; Bray, F. (Eds.) The Cancer Atlas, 3rd ed.; American Cancer Society: Atlanta, GA, USA, 2019; Available online: http://www.cancer.org/canceratlas (accessed on 2 November 2022).
- Gupta, A.K.; Galoforo, S.S.; Berns, C.M.; Martinez, A.A.; Guan, K.L.; Lee, Y.J. Elevated levels of ERK2 in human breast carcinoma MCF-7 cells transfected with protein kinase Cα. Cell Prolif. 1996, 29, 655–663.
- Varga, A.; Czifra, A.; Tállai, B.; Németh, T.; Kovács, I.; Kovács, L.; Bıró, T. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur. Urol. 2004, 46, 462–465.
- Kho, D.H.; Bae, J.A.; Lee, J.H.; Cho, H.J.; Cho, S.H.; Seo, Y.-W.; Ahn, K.Y.; Chung, I.J.; Kim, K.K. KITENIN recruits Dishevelled/PKCd to form a functional complex and controls the migration and invasiveness of colorectal cancer cells. Gut 2009, 58, 509–519.
- Lee, M.-S.; Kim, T.Y.; Kim, Y.-B.; Lee, S.-Y.; Ko, S.-G.; Jong, H.-S.; Bang, Y.-J.; Lee, J.W. The Signaling Network of Transforming Growth Factor β1, Protein Kinase Cδ, and Integrin Underlies the Spreading and Invasiveness of Gastric Carcinoma Cells. Mol. Cell. Biol. 2005, 25, 6921–6936.
- Mandil, R.; Ashkenazi, E.; Blass, M.; Kronfeld, I.; Kazimirsky, G.; Rosenthal, G.; Umansky, F.; Lorenzo, P.S.; Blumberg, P.M.; Brodie, C. Protein kinase Cα and protein kinase Cδ play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001, 61, 4612–4619.
- Kim, M.; Kim, R.; Yoon, C.; An, S.; Hwang, S.-G.; Suh, Y.; Park, M.-J.; Chung, H.Y.; Kim, I.G.; Lee, S.-J. Importance of PKCδ signaling in fractionated radiation-induced expansion of glioma-initiating cells and resistance to cancer treatment. J. Cell Sci. 2011, 124, 3084–3094.
- Yang, Y.-L.; Chu, J.-Y.; Luo, M.-L.; Wu, Y.-P.; Zhang, Y.; Feng, Y.-B.; Shi, Z.-Z.; Xu, X.; Han, Y.-L.; Cai, Y.; et al. Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer 2008, 47, 127–136.
- Liu, S.-G.; Wang, B.-S.; Jiang, Y.-Y.; Zhang, T.-T.; Shi, Z.-Z.; Yang, Y.; Yang, Y.-L.; Wang, X.-C.; Lin, D.-C.; Zhang, Y.; et al. Atypical protein kinase Cι (PKCι) promotes metastasis of esophageal squamous cell carcinoma by enhancing resistance to anoikis via PKCι-SKP2-AKT Pathway. Mol. Cancer Res. 2011, 9, 390–402.
- Lahn, M.; Su, C.; Li, S.; Chedid, M.; Hanna, K.R.; Graff, J.R.; Sandusky, G.E.; Ma, D.; Niyikiza, C.; Sundell, K.L.; et al. Expression levels of protein kinase C-α in non-small-cell lung cancer. Clin. Lung Cancer 2004, 6, 184–189.
- Bae, K.; Wang, H.; Jiang, G.; Chen, M.G.; Lu, L.; Xiao, L. Protein kinase Cε is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res. 2007, 67, 6053–6063.
- Oka, M.; Kikkawa, U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005, 24, 287–300.
- la Porta, C.A.M.; di Dio, A.; Porro, D.; Comolli, R. Overexpression of novel protein kinase C δ in BL6 murine melanoma cells inhibits the proliferative capacity in vitro but enhances the metastatic potential in vivo. Melanoma Res. 2000, 10, 93–102.
- Abrams, S.T.; Lakum, T.; Lin, K.; Jones, G.M.; Treweeke, A.T.; Farahani, M.; Hughes, M.; Zuzel, M.; Slupsky, J.R. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CβII. Blood 2007, 109, 1193–1201.
- Holler, C.; Piñón, J.D.; Denk, U.; Heyder, C.; Hofbauer, S.; Greil, R.; Egle, A. PKCβ is essential for the development of chronic lymphocytic leukemia in the TCL1 transgenic mouse model: Validation of PKCβ as a therapeutic target in chronic lymphocytic leukemia. Blood 2009, 113, 2791–2794.
- Kabir, N.N.; Rönnstrand, L.; Kazi, J.U. Protein kinase C expression is deregulated in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 2288–2290.
- Espinosa, I.; Briones, J.; Bordes, R.; Brunet, S.; Martino, R.; Sureda, A.; Prat, J.; Sierra, J. Membrane PKC-beta 2 protein expression predicts for poor response to chemotherapy and survival in patients with diffuse large B-cell lymphoma. Ann. Hematol. 2006, 85, 597–603.
- Nicolle, A.; Zhang, Y.; Belguise, K. The emerging function of PKC theta in cancer. Biomolecules 2021, 11, 221.
- Page, A.; Navarro, M.; Suarez-Cabrera, C.; Bravo, A.; Ramirez, A. Context-dependent role of IKKß in cancer. Genes 2017, 8, 376.
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158.
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-ķß and Ikß proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52.
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324.
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254.
- Lawlor, M.; Alessi, D.R. PKB/AKT: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910.
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657.
- Revathidevi, S.; Munirajan, A. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91.
- Li, W.; Liang, X.; Zeng, Z.; Yu, K.; Zhan, S.; Su, Q.; Yan, Y.; Mansai, H.; Qiao, W.; Yang, Q.; et al. Simvastatin inhibits glucose uptake activity and GLUT4 translocation through suppression of the IR/IRS-1/Akt signaling in C2C12 myotubes. Biomed. Pharmacother. 2016, 83, 194–200.
- Zhang, C.; Yang, N.; Yang, C.H.; Ding, H.S.; Luo, C.; Zhang, Y.; Wu, M.-J.; Zhang, X.-W.; Shen, X.; Jiang, H.-L.; et al. A novel anticancer agent, exerts its anti-proliferative activity by interfering with both PI3K-Akt-mTOR signaling and microtubule cytoskeleton. PLoS ONE 2009, 4, e4881.
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B.; Martinez-Lostao, L.; de Miguel, D.; et al. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 10, 1603–1624.
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475.
- Li, Z.; Zhang, L.; Liu, D.; Wang, C. Ceramide glycosylation and related enzymes in cancer signaling and therapy. Biomed. Pharmacother. 2021, 139, 111565.
- Neshat, S.; Rezaei, A.; Farid, A.; Sarallah, R.; Javanshir, S.; Ahmadian, S.; Chatrnour, G.; Daneii, P. The tangled web of dyslipidemia and cancer: Is there any association? J. Res. Med. Sci. 2022, 27, 93.
- Ho, J.; Kim, E.; Han, M.; Jung, I.; Lee, J.; Suk Jo, Y. Impact of Dyslipidemia on the Risk of Second Cancer in Thyroid Cancer Patients: A Korean National Cohort Study. Ann. Surg. Oncol. 2021, 28, 4373–4384.
- Ferreira, L.M. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Garcia-Heredia, J.M.; Carnero, A. Decoding Warburg’s hypothesis: Tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 2015, 6, 41582–41599.
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 20, 297–308.
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664.
- Zielonka, J.; Kalyanaraman, B. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis—A critical commentary. Free Rad. Biol. Med. 2008, 45, 1217–1719.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92.
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol.-Cell Physiol. 2015, 309, C569–C579.
- Hagen, T.; Vidal-Puig, A. Mitochondrial uncoupling proteins in human physiology and disease. Minerva Medica 2002, 93, 41–57.
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cardenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99.
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Huang, J.; Wang, G.; Liao, K.; Xie, N.; Deng, K. UCP1 modulates immune infiltration level and survival outcome in ovarian cancer patients. J. Ovarian Res. 2022, 15, 16.
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 21, 960–965.
- Li, W.; Zhang, C.; Jackson, K.; Shen, X.; Jin, R.; Li, G.; Kevil, C.G.; Gu, X.; Shi, R.; Zhao, Y. UCP2 knockout suppresses mouse skin carcinogenesis. Cancer Prev. Res. 2015, 8, 487–491.
- Jezek, J.; Jaburek, M.; Zelenka, J.; Jezek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747.
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581.
- Drochiolu, G. Multifactorial distress, the Warburg Effect, and respiratory and pH imbalance in cancer development. Stresses 2023, 3, 500–528.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 13, 735.
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Xia, T.; Chen, D.; Piao, H.-l.; Liu, H.-X. The double-edged role of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857.
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2017, 313, R646–R653.
- Lee, J.; Roh, J.-L. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023, 559, 216119.
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.-y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103.
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22.
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191.
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228.
- The α-Tocopherol, ß-Carotene Cancer Prevention Study Group. The effect of vitamin E and ß-carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1996, 330, 1029–1035.
- Black, H.S.; Boehm, F.; Edge, R.; Truscott, T.G. The benefits and risks of certain dietary carotenoids that exhibit both anti- and pro-oxidative mechanisms—A comprehensive review. Antioxidants 2020, 9, 264.
- Szkudlinska, M.A.; von Frankenberg, A.D.; Utzschneider, K.M. The antioxidant N-Acetylcysteine does not improve glucose tolerance or ß-cell function in type 2 diabetes. J. Diabetes Its Complicat. 2016, 30, 618–622.
- Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) supplementation improves impaired mitochondrial fuel oxidation and lowers insulin resistance in patients with type 2 diabetes: Results of a pilot study. Antioxidants 2022, 11, 154.
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857.
- Hibino, M.; Maeki, M.; Tokeshi, M.; Ishitsuka, Y.; Harashima, H.; Yamada, Y. A system that delivers an antioxidant to mitochondria for the treatment of drug-induced liver injury. Nat. Sci. Rep. 2023, 13, 6961.

