1. Diabetic Metabolism Linking Cancer
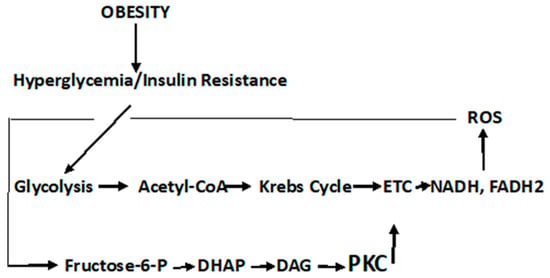
It should be noted that the pathophysiological responses in T2DM are complex and both cell- and tissue-specific. Likewise, the carcinogenic process is equally complex and also cell- and tissue-specific. Recognizing the complexity of these two diseases, the description of diabetic metabolism are synthesized herein, to present a general, comprehensive overview. It is imperative that the role of oxidative stress and ROS formation in diabetes be described, as the confluence of these diabetic responses to ROS sets the background linking this disorder to cancer. In this regard, persistent hyperglycemia leads to repeated, acute changes in cellular metabolism initiating four metabolic pathways induced through ROS production that, in turn, lead to higher levels of ROS and oxidative stress. These pathways are activated by increased ROS levels that elevate PARP ((poly (ADP-ribose) polymerase)) levels and downregulate GADPH (glyceraldehyde-3-phosphate dehydrogenase) levels. The latter activate the Polyol pathway (), the Hexosamine pathway ()] the Protein Kinase C pathway (
Figure 3), and the formation of advanced glycation products [
28,
78,
86,
87,
91].
Scheme 1. Polyol Pathway. It is estimated that under persistent hyperglycemic conditions, the Polyol pathway is activated by elevated ROS levels that, in turn, elevate PARP levels and downregulate GADPH levels. The latter activate and upregulate the Polyol and Hexosamine pathways, accelerate the formation of advanced glycation products (AGE), and activate the Protein Kinase C signal transduction pathway (PKC)—all of which lead to further ROS production, tissue damage, and diabetic complications.
Scheme 2. Hexosamine Pathway. GFAT, glutamine: fructose-6-phosphate-aminotransferase; UDP-GlucNAc, Uridine-5-diphosphate-N-acetylglucosamine; GlutN, glutamine; Glu, glutamic acid; UDP, Uridine 5′-diphosphate; Pi, inorganic phosphate.
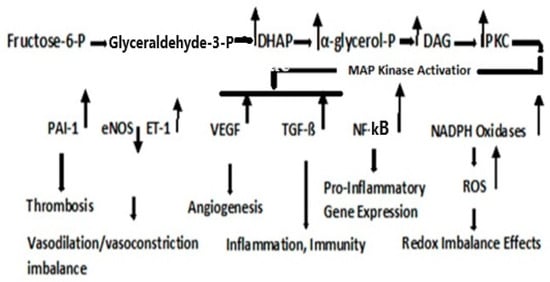
Figure 3. Protein Kinase C Pathway. Metabolism of fructose-6-phosphate results in the upregulation in dihydroxyacetone phosphate (DHAP), α-glycerol-phosphate, and diacylglycerol (DAG). PKC activation, although cell type- and isoform-specific, is generally activated via DAG; although the flux through the Hexosamine pathway, regulated by GFAT, may also be involved in the activation of PKC [
87].
As the high levels of intracellular glucose are reduced by aldose reductase to sorbitol, the reaction consumes NADPH, a cofactor critical for the regeneration of the natural antioxidant, reduced glutathione (refer to
Figure 1). Decreasing the level of reduced glutathione increases intracellular oxidative stress [
86]. Two NAD+ degradative reactions, a mitochondrial family of signaling proteins (Sirtuins), a histone deacetylase, and an ADP ribosyl transferase are NAD+ dependent, the latter consumed in the formation of nicotinamide, thus contributing to the NADH/NAD redox imbalance [
92,
93]. Thus, the Polyol pathway plays a critical part in pathophysiology, which contributes to diabetic complications and is initiated by the formation of ROS.
Fructose-6-phosphate is metabolized either through the Embden–Meyerhof–Parnas pathway, or under conditions when the rate of the reoxidation of NADPH is limited, and may be produced when the glucose metabolism is diverted through the pentose phosphate shunt. Nevertheless, fructose-6-phosphate is the initial point of both the Hexosamine and PKC pathways. The Hexosamine Pathway is depicted in [
28].
GFAT regulates the flux through the Hexosamine pathway and is involved in the etiology of diabetic nephropathy [
94]. Uridine-5-diphosphate-N-acetylglucosamine (UDP-GlucNAc) is the precursor of amino sugars required for the synthesis of proteoglycans, glycolipids, and glycoproteins.
The PKC pathway is a signaling pathway that not only results in pathophysiological manifestations of T2DM but is also a direct link to cancer.
2. Dyslipidemia and Cell Signaling
It is known that hyperglycemia, as it occurs in T2DM, results in an elevation of circulating triglycerides and free fatty acids (FFAs). This condition denotes serious dysfunction in lipid dynamics and leads to severe diabetic clinical complications [
112]. Dyslipidemia is also a major risk factor for cardiovascular disease (CVD) [
113]. Indeed, CVD and cancer share several similar risk factors. e.g., obesity, T2DM, dyslipidemia, chronic inflammation, oxidative stress, and cytokine production—all mediators that contribute to the connection of T2DM/CVD and cancer [
113,
114,
115,
116,
117,
118]. Although conflicting evidence of CVD and cancer lipidomic risk profiles have been observed [
119], other studies have demonstrated that elevated levels of lipid biomarkers are independently associated with all-cause mortality as well as CVD risk [
118]. Further evidence of a CVD and cancer link was found when study participants who met 6–7 of Life’s simple seven ideal health ASCV (atherosclerotic cardiovascular) metrics [
119], exhibited a 51% lower risk for cancer incidence, and those at high CV risk exhibited a >3-fold increased risk of cancer, compared with low-CV risk subjects [
119]. Thus, a link runs from T2DM to CVD to cancer, although the complexity of this link is multifaceted.
Type 2 Diabetes Mellitus-associated dyslipidemia may partially be a consequence of a systemic FFA flux secondary to insulin resistance [
120,
121]. At least 35% of gluconeogenesis in T2DM patients is FFA dependent. Efforts to explain the competitive oxidation between glucose and FFA resulted in what became known as the Glucose Fatty-Acid cycle, or the Randle cycle [
122]. Fatty acids are first transported across the cell membrane by Fatty Acid Transport Protein 1 (FATP1), where an FA acyl-CoA synthetase yields acetyl- CoA in the cytosol or a carnitine palmitoyl transfers acyl-CoA across the mitochondrial membrane. A major convergence of signaling pathways involves Protein Kinase C (PKC) activation. Under hyperglycemic conditions in T2DM, incoming surplus energy from obesity is stored in adipocytes in the form of lipids or triacylglyceride (TAG). TAG is converted to diacylglyceride (DAG) by triglyceride lipase (TGL). An upregulation in DAG occurs through the metabolism of Fructose-6-P (
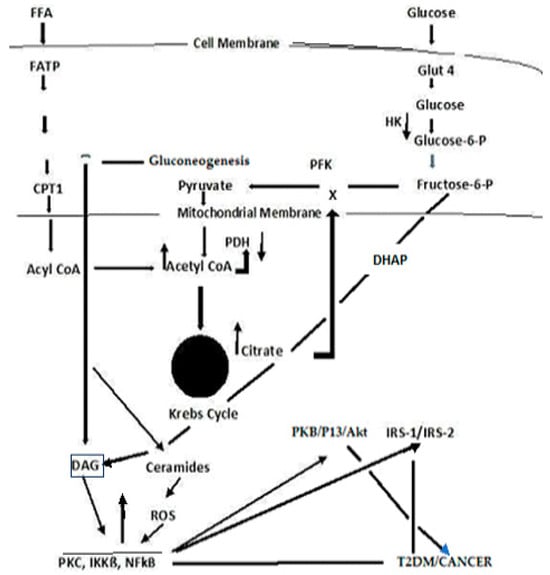
Figure 3). The convergence of these pathways to produce DAG is depicted in
Figure 4. PKC upregulation is generally activated by DAG [
95]
Figure 4. A simplified schema depicting the confluence of glucose and free fatty acid ROS-induced signaling that links T2DM and cancer.
Although the Randle cycle has been discussed previously, with respect to T2DM [
28], a recapitulation is necessary to place the importance of these reactions in perspective to the link between T2DM and cancer. Acyl-CoA formation, in both the cytosol and mitochondria, induces ß-oxidation of FAs [
123,
124,
125]. In the mitochondria, this results in the formation of Acetyl-CoA, which feeds into the Krebs cycle, increasing citrate that, in turn, inhibits hexokinase and pyruvate dehydrogenase activities. The inhibition of hexokinase results in the downregulation of glucogenesis. Elevated Krebs cycle activity results in an increased formation of FADH
2 and NADH that yield ROS as electrons that are passed down the ETC [
125]. The Acyl-CoA formed in the cytosol stimulates ß-oxidation in the peroxisome and upregulates the glyoxylate cycle by which glucose is synthesized.. DAG activates PKC, IKKB, and NF-kß; the latter acts to suppress PKB, P13/Akt, and IRS-1 and 2. The inhibition of PKB function enhances T2DM, whereas the gain of function drives cancer. The deregulation of P13 plays an important role in proliferation and tumorigenesis. Akt protein plays a critical role in cell survival and the epithelial–mesenchymal transition for cancer formation.
A ROS attack on PUFA leads to increased lipid peroxidation that aggravates systemic inflammation [
126]. The relationship between inflammation and cancer was recognized in the mid-1800s when Virchow theorized that cancers originated at sites of chronic inflammation. The causal relationship between inflammation, innate immunity, and cancer is now recognized [
127,
128,
129,
130]. Cytokines, and this includes “chemokines”, which are chemotactic cytokines, are the messengers for most of the biologic effects of the immune system, e.g., cell-mediated immunity and allergic responses [
128]. The major source of cytokines/chemokines are T lymphocytes. Chemokines play a central role in the development and homeostasis of the immune system and are involved in all protective or destructive immune and inflammatory responses [
129,
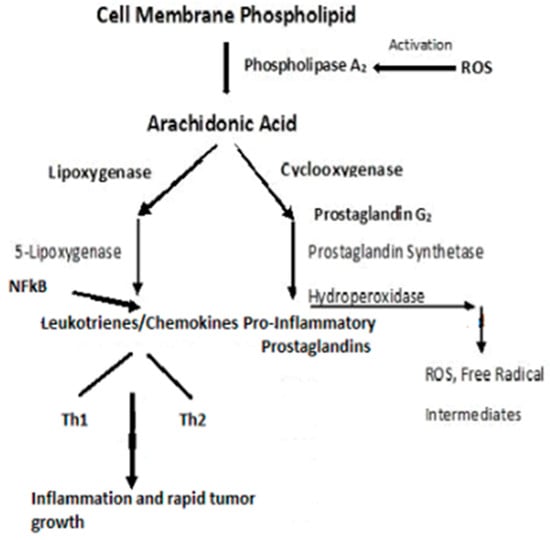
130]. T lymphocytes are characterized by the presence of cell surface molecules, CD4 or CD8. Those lymphocytes expressing CD4 are known as helper T-cells and are prolific cytokine producers and are further subdivided into subsets Th1 and Th2. Th1 produces pro-inflammatory responses. The pro-inflammatory cytokine, Tumor Necrosis Factor-α, (TNF-α), a proinflammatory cytokine—regulates inflammatory cell populations, but once homoeostasis is imbalanced and both Th1 and Th2 arms produce an overabundance of proinflammatory cytokines, rapid tumor growth and proliferation occur [
128]. (
Figure 5). Indeed, in studies on particulate lung carcinogenesis, it has been shown that chronic inflammation alone can initiate tumor growth without direct interaction with DNA [
131].
Figure 5. Pro-inflammatory schema leading to inflammation and rapid tumor growth.
PKC isozymes, phospholipid-dependent serine/threonine kinases, signal through multiple transduction pathways and, in cancer cells, are implicated in angiogenesis, cell proliferation, tumor promotion, invasion, migration, metastasis, and apoptosis (cell survival) [
132,
133,
134]. The PKCs’ role in normal cell function and that in cancer are complicated in that, based upon their structural and activation properties, three subfamilies are classified as classic PKC isozymes that require DAG as an activator and Ca
2+ as a cofactor; non-classic, regulated by DAG with no Ca
2+ required; and atypical PKC that is not activated by DAG [
135]. Thus, the various isoforms may exhibit different responses dependent upon their target proteins and their subsequent signaling responses, with some isoforms even acting as tumor suppressors [
134]. Generally, however, it is recognized that PKCs are associated with a number of types of cancer, including breast [
11,
136], bladder [
137], colon [
138], gastric [
139], glioma [
140,
141], head and neck [
142,
143], lung [
144,
145], melanoma [
146,
147], and some types of leukemia [
148,
149,
150,
151]. The most widely studied isozymes, in relation to cancer, have been PKC α, β, ε, and δ [
134]. The theta isozyme, PKCѲ, is classified in a novel PKC subfamily, and its expression is limited to only a few cell types. However, it controls T-cell activation, survival, and differentiation [
152]. PKCѲ is highly expressed in T-cell immune responses, playing a critical role for the T-helper (Th2 and Th17)-mediated responses, while the cytotoxic T-cell-driven responses remain relatively intact. Although the function and mode of action of this isoform are different, depending on the type of cancer, in most cancers, the presence of an elevated PKCѲ leads to an abnormal proliferation, migration, and invasion of cancer cells and, thus, promotes tumor aggressiveness [
152].
IKKß (inhibitory kß phosphorylase) is a serine/threonine kinase that is upregulated either through the conversion of triacylglyceride (TAG) to diacylglyceride (DAG) in the FFA arm of the Randle cycle or upregulation in DAG through the metabolism of Fructose-6-P in the glucose arm of the cycle [
153,
154] (
Figure 4). IKKß phosphorylates and deregulates NF-ķß, a serine/threonine kinase nuclear transcription factor that has critical roles in inflammation, immunity, cell proliferation, and apoptosis [
153,
154,
155,
156]. NF-ķß may also be activated by proinflammatory cytokines, e.g., Tumor Necrosis Factor (TNF)-α [
156]. Most known hallmarks of cancer are affected by NF-ķß activation [
156]. Targeting IKKß, via inhibitors, may provide therapeutic opportunities [
154].
PKC activation induces a cascade of kinases, including mitogen-activated serine/threonine proteins (MAP Kinases) that regulate cell differentiation, proliferation, and apoptosis [
156]. Although the downstream secondary signaling cascades are numerous and complex, one, depicted in
Figure 4, has particular importance with respect to T2DM and cancer, i.e., the NF-ķß suppression of IRS, PKB/Akt, Serine/threonine-based proteins, protein kinase B (PKB), are also known as Ant, as the widely expressed isoforms of PKB; PKB α, ß, and γ are also known as Ant1, Ant2, and Ant3 [
157,
158,
159,
160].
IRS-1, Insulin Receptor Substrate, phosphorylation and dysregulation reduce GLUT4 translocation to the cell surface, decreasing insulin-stimulated glucose transport and glucose uptake. IRS-1 also disrupts subsequent cell signaling pathways, contributing to the development of T2DM [
161,
162]. NF-ķß targets P13 (Phosphatidylinositol 3-kinase), which plays a central role in a complex, multi-armed signaling network that orchestrates cell responses, including cell survival, growth, proliferation, angiogenesis, migration, and glucose metabolism [
157,
158]. PI3K is presumed to activate most of its downstream targets via Akt, a serine/threonine kinase, that affects the aforementioned cellular responses and may provide a therapeutic target [
163,
164,
165].
Contributing to an already intricate and complex picture, is the formation of ceramides [
164,
165] (
Figure 4). Ceramide is a core sphingolipid and generally produces antiproliferative responses, e.g., cell growth inhibition, apoptosis induction, and cell invasiveness—thus acting as a tumor suppressor. However, the ceramide metabolism involves glycosylation to produce an AGE that reacts with receptor sites to form a RAGE that, in turn, releases ROS. The tumor suppressor function is lost, and ROS activate the PKC signal transduction pathway. Ceramide glycosylation is closely linked to drug resistance, and this has become a high interest area of investigation for therapeutic targets for cancer [
165].
In summary, dyslipidemia is a hallmark of T2DM and cancer [
28,
112,
123]. Dyslipidemia has also been associated with a range of cancers, including secondary cancers, e.g., colon, liver, pancreas, ovary, prostate, kidney, bladder, and brain cancers [
166,
167].
3. Cancer Cell Metabolism, ROS, and Antioxidant Therapy
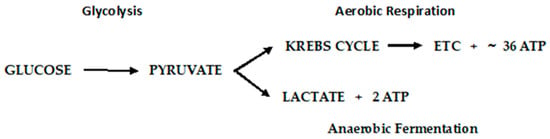
Otto Warburg first observed, in 1922, that cancer cells exhibit a specific metabolic pattern—one characterized by a shift from aerobic respiration to anaerobic fermentation (the Warburg Effect) [
168]. The aerobic respiratory metabolic pattern of normal cells and anaerobic fermentation of cancer cells are depicted in
Figure 6.
Figure 6. Metabolic pathways of glucose metabolism of normal (aerobic respiration) and cancer cells lactic acid fermentation).
The Warburg Effect raises a number of questions that have been systematically addressed [
169]. First, how does a switch to a much lower yielding ATP pathway as fermentation sustain tumor growth? Using mouse ascites (cancer) cells that obtain ~100% of their energy from fermentation, it was determined that normal mouse cells consumed an average of seven mm
3 of oxygen.mg/h., whereas fermentation produced 60 mm
3 of lactic acid/mg/h. Converted to energy equivalents, cancer cells obtain approximately the same amount of energy from fermentation as normal cells do through aerobic respiration. A second question, recognizing that the respiration of all cancer cells is irreversibly damaged (irreversible damage occurs as the restoration of oxygen does not restore cells’ normal respiration), is how this damage is induced without killing the cells? Warburg postulated that damage to the respiratory system could be induced by decreasing the oxygen consumption, consequently decreasing the yield of ATP, or the uncoupling of respiration and ATP production with undiminished oxygen consumption. Injury to respiration will be irreversible, and this is common to all cancer incitants. The calculation of metabolic quotients demonstrated that the first phase of carcinogenesis (the irreversible damaging of respiration) need not involve a decrease in the respiratory quotient but entail the uncoupling of oxidative phosphorylation without undiminished oxygen consumption. Warburg provided striking confirmation of his main conclusions from metabolic studies on the C
3H/He mouse cell lines developed at the NCI. Two cell lines, developed from a single cell, demonstrate a high and low malignancy rate when injected into C
3H/He mice. The anaerobic glycolysis quotient for the high malignancy line was Q
MN2 = 60–80; that of the low malignancy rate was 20–30. The aerobic glycolysis values, Q
MO2, was 30 vs. 10 for the high and low malignancy lines, respectively. They were of lower magnitude because of the Pasteur Effect, which was greater in the high malignancy cell line. The Pasteur Effect is the inhibiting effect of oxygen upon fermentation. As oxygen is increased, the accumulation of fermentation products is repressed, and there is a decline in the rate of carbohydrate dissimilation. Conversely, the rate of oxygen consumption (Q
O2 = 5–10) in the high malignancy line was less than that of the low malignancy line (Q
O2 = 10–15), corresponding to a greater level of respiratory damage in the high malignancy line. In toto, there is strong evidence consistent with the Warburg Effect, but the question of how this irreversible damage is induced remains open and a very active area of investigation.
Some critics of Warburg’s hypothesis, i.e., that the “driver of tumorigenesis is defective cellular respiration” have proposed alternative possibilities, particularly with the activation of oncogenes and inactivation of tumor suppressor genes [
168,
169,
170]. It is posited that damaged mitochondria are not the root cause of the aerobic glycolytic lesion exhibited by most tumor cells but result from oncogene-directed metabolic reprogramming required to support anabolic growth [
171]. It is argued that most tumor mitochondria are not defective in their ability to conduct oxidative phosphorylation and that anabolic growth is the result of oncogene-directed metabolic programming and that the metabolites can be oncogenic by altering cell signaling and blocking cellular differentiation. In this scenario, the activation of the P13/Akt pathway leads to enhanced glucose uptake, glycolysis, increased glucose transporter expression, and the activation of hexokinase. Increased nutrient intake, glucose and glutamine, supports the anabolic requirements of cell growth, whereas proliferating cells use strategies to decrease their ATP production. The overall hypothesis is that the reprogramming of the cell’s metabolism toward macromolecular synthesis is critical for maintaining cell mass and reaching G
2 phase in preparation for cell division. In this reprogrammed metabolism, the need is greater for reduced carbon and nitrogen and NADPH for reductive biosynthetic reactions. However, with respect to a link between T2DM and cancer, it should be noted that hyperglycemia leads to the activation of the Hexosamine pathway, which would limit glutamine availability [
28]. Further, in insulin-resistant cells, mitochondrial respiration, glycolysis, and ATP levels decrease (in part due to changes in the glucose transporter (GLUT4)—all conditions associated with cancer cells.
Seeming contradictory to the argument that most tumor mitochondria are not defective in their ability to carry-out oxidative phosphorylation, control of the latter’s metabolic machinery resides in the mitochondrial DNA (mtDNA), and there have been extensive studies conducted to examine the mitochondrial genome [
172,
173]. It is posited that long-term consequences induced by ROS are the results of alterations in mtDNA and indeed, mutations in Complex 1, ubiquinone oxidoreductase, of the ETC are derived from mutations in mtDNA [
172]. Although mutations in mtDNA occur at high frequency, the question of whether these mutations alter tumor behavior has been difficult to discern. Using the mtDNA from two tumor cell lines, one highly metastatic, the other of low metastatic potential, the transfer of the mtDNA into recipient tumor cells conveyed the metastatic potential of the transferred mtDNA. The mutations produced a deficiency in respiratory Complex I and produced an overabundance of ROS. Experimental results indicated that mtDNA mutations contributed to tumor progression by enhancing the metastatic potential of tumor cells [
172]. A commentary to this study suggested that, using the methodology employed, the researchers failed to show evidence for the formation of superoxide and hydrogen peroxide that was presumed to be generated from the Complex I deficiency associated with mtDNA mutations [
173]. Nevertheless, all of the mutations that affect Complex I have similar consequences, i.e., they promote an increase in ROS, increase succinate, inhibit mitochondrial pyruvate dehydrogenase (reducing the flux of pyruvate into the Krebs cycle), and stabilize Hypoxia-Inducing Factor 1-α (HIF-1α) [
174].
Hypoxia (low oxygen tension) is thought to be one of the main elements in the switch between glycolysis and respiration [
174,
175]. Hypoxia induces a complex of intracellular signaling pathways including P13/Akt, MAPK, NFkß, and HIF—all of which are involved in cell proliferation, apoptosis, glucose metabolism, metastasis, and inflammation [
175]. Low-oxygen availability inhibits oxidative phosphorylation. The adaption of a cell to hypoxia is partially dependent on the expression and stabilization of Hypoxia-Inducing Factor 1-α (HIF-1α), a transcription protein that when overexpressed, is implicated in promoting tumor growth and metastasis. The overexpression of HIF-1α in tumor cells and rapidly growing normal cells stimulates glycolysis and restricts mitochondrial respiration. The inadequate regulation of hypoxia is an important contributor to the malignant phenotype. Hypoxia also leads to immune resistance and immune suppression that aid tumor cells to escape immune surveillance [
176].
Considering the second possibility for the transition to the Warburg phenotype, i.e., the uncoupling of respiration and oxidative phosphorylation, mitochondrial uncoupling proteins (UCPs) catalyze a regulated proton leak across the inner mitochondrial membrane without the generation of ATP [
177]. ROS (superoxide) and long-chain fatty acids activate UCP-1, which can be inhibited by purine nucleotides, e.g., ATP [
178,
179]. There are five isoforms of UCP that have been identified thus far, each with specific functions [
180]. An increased expression of UCP-1 has been shown to play a relevant role in immune infiltration by regulating oncogene levels in ovarian cancer [
180]. Based on cancer single-cell sequencing data, tumor functional status analyses suggest that UCP-1 may down regulate invasion, epithelial-mesenchymal transition, metastasis, DNA repair, and angiogenesis. UCP-2 inhibits ROS production, which results in a reduced ADP yield and reduced insulin secretion. UCP-2 also catalyzes the exchange and transport of intramitochondrial C4 intermediates (e.g., oxaloacetate), which negatively controls the oxidation of acetyl-CoA-producing substrates through the Krebs cycle. This lowers the redox pressure on the mitochondrial respiratory chain, the ATP–ADP ratio, and ROS production [
181]. Employing a UCP-2 knockout mouse, the first in vivo evidence reported that UCP-2 significantly reduced the chemically induced formation of papilloma and malignant squamous cell carcinomas of the skin while not affecting apoptosis [
182]. Lactate generation was significantly increased in the carcinogen-treated wild-type mice, but there was no difference between carcinogen-treated and vehicle-treated UCP-2 knockout mice. An upregulation in UCP-2 is known to promote aerobic glycolysis and increase lactate levels. Mitochondrial phospholipase A
2, activated by ROS, induces uncoupling in heart mitochondria [
183].
Seeking the “switch” that turns normal cells into the cancer phenotype seems inexpedient considering the cacophony of biological responses that may occur simultaneously within an indeterminate time span. As an example, consider cell signaling pathways, including P13/Akt, MAPK, NFkß, and HIF, and their myriad of secondary responses; oxygen tension and the overexpression and stabilization of HIF-1α; mutations in mtDNA, especially those affecting complexes in the respiratory chain; and the uncoupling of respiration from oxidative phosphorylation, are all involved in the reprogramming of the metabolism of the cell [
171,
184]. If there is a single “switch”, ROS must be considered the prime candidate. Indeed, while recognizing that cancer initiation may be due to multifactorial stressors, evidence suggests a close relationship between oxidative stress and carcinogenesis [
185,
186]. In addition, ROS not only leads to redox imbalance but functions as signaling molecules that activate signal transduction pathways that promote many aspects of tumor development and progression, expressed in the cancer phenotype. This is particularly relevant in the case of obesity-related hyperglycemia in T2DM, where elevated ROS are produced, and these act as signal molecules to initiate signal transduction pathways ().
Scheme 3. Hyperglycemic-induced ROS that activates signal transduction pathways.
Through a somewhat tortuous journey of possibilities, the conclusion remains the same for the initiation of the cancer phenotype, i.e., “reprogrammed metabolism should now be considered as a core hallmark of cancer” [
113,
171], and cancer joins T2DM, Metabolic Syndrome, and CVD, as a metabolic disease.
4. ROS’ Role in the Early and Late Stages of Cancer
Mitochondrial damage and the resulting dysfunction of respiratory components that are induced by ROS, lead to malignant transformations [
35]. These organelles are essential for energy metabolism, apoptosis regulation, and cell signaling [
186]. The overproduction of ROS, as it occurs in hyperglycemia, induces cancer development by causing genomic instability (mtDNA), inducing signal transduction pathways that modify gene expression (tumor proto-oncogenes and tumor suppressor genes), and impairs oxidative phosphorylation. The latter results in the production of more ROS, which aggravates physiological and metabolic dysfunctions. These elevated levels of ROS can cause cell apoptosis. ROS levels critically determine whether ROS augment tumorigenesis or terminate tumorigenesis via apoptosis [
29]. Thus, a homeostatic balance of ROS must be maintained for normal cell survival [
29,
35,
186].
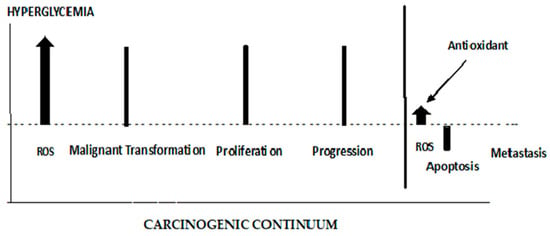
The role of ROS in initiating the early stages of carcinogenesis is depicted in
Figure 7. ROS are elevated from the Hyperglycemic metabolism. Elevated ROS results in malignant cell transformations. High ROS levels can lead to apoptosis; therefore, any effective antioxidant therapy should be administered prior to cancer cell proliferation. When antioxidants are administered late in the carcinogenic continuum, ROS levels are suppressed, and apoptosis is lowered. The cancer cells lose their polarity, cell–cell adhesion, and gain mobility. This epithelial to mesenchymal transition is a major cause of tumor metastasis [
187,
188]. As seen in
Figure 7, antioxidant therapy at this late stage of carcinogenesis results in a boost in metastasis. It has been demonstrated that increased activity (decreased stress) of the GPX4 (a selenocysteine-containing protein pathway), known to be regulated via redox homeostasis, is causally linked to the enhanced metastatic activity of cancer cells [
187]. Dysregulated cholesterol homeostasis also results in resistance to ferroptosis, which results in increased tumorigenesis and metastasis [
188]. A dietary supplementation of antioxidants or induced glutathione synthesis can increase GPX4 activity and promote distant metastasis in animal models of lung cancer and melanoma [
189,
190]. Thus, the timing of antioxidant therapy is obviously important and should be initiated as early as possible. If dietary interventions are not successful in addressing dietary risks leading to hyperglycemia, the antioxidant therapy should begin in the pre-diabetic phase of T2DM. The discontinuation of antioxidant therapy should take place before the later stages of carcinogenesis and metastasis. Unfortunately, up to this point, antioxidant supplementation, in clinical trials, in order to maintain or restore redox balance and prevent cancer, have largely either failed or been controversial for reasons given previously [
42,
43,
44]. GlyNAC has shown some promise when it is shown to avoid the detrimental effects of NAC supplementation [
76,
77,
78,
79]. A recent report describes a mitochondrion-targeting nanoparticle that delivers an antioxidant directly to mitochondria, the major source of endogenous ROS [
191]. The perfection of these targeting systems hold promise for targeted antioxidant therapy.
Figure 7. The different roles of ROS and antioxidants in the early and late stages of cancer.
This entry is adapted from the peer-reviewed paper 10.3390/jmp5010007