 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salvatore Cortellino | -- | 8317 | 2024-03-06 07:52:56 | | | |
| 2 | Lindsay Dong | Meta information modification | 8317 | 2024-03-07 01:50:50 | | |
Video Upload Options
Inflammation is a key contributor to both the initiation and progression of tumors, and it can be triggered by genetic instability within tumors, as well as by lifestyle and dietary factors. The inflammatory response plays a critical role in the genetic and epigenetic reprogramming of tumor cells, as well as in the cells that comprise the tumor microenvironment. Cells in the microenvironment acquire a phenotype that promotes immune evasion, progression, and metastasis.
1. Introduction
2. Inflammation
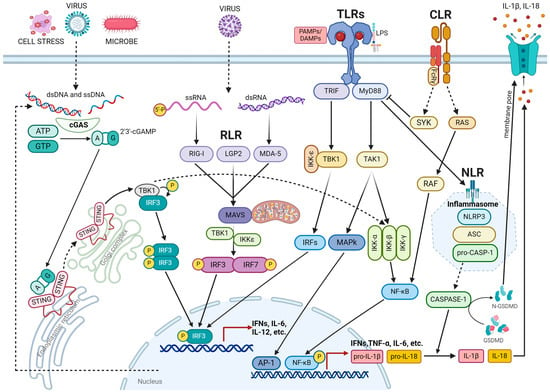
3. DNA Damage Response in Inflammation
3.1. Mechanism Underlying DNA Damage-Induced Inflammation
3.2. DNA Repair Deficiency Disorder and Inflammation
3.3. Inflammation Promoted by Transposable Element (TE) Instability
3.4. Inflammation Triggered by R-Loops Resolving Defects
3.5. Anti-Tumor Cytotoxicity Mediated by Inflammatory Response
4. Epigenetics and Inflammation
4.1. Epigenetic Regulatory Mechanisms
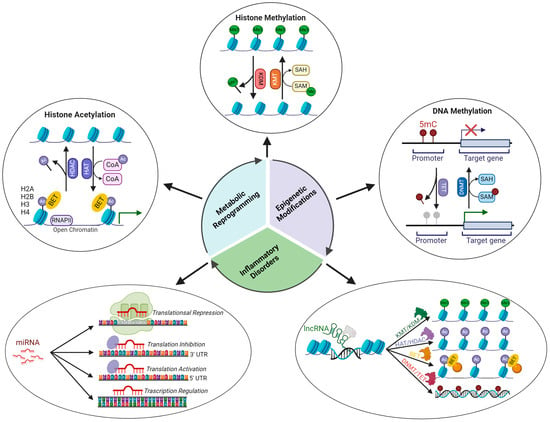
4.2. Epigenetic Signatures Underlying Inflammation
Histone acetylation. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate the expression of several genes involved in the inflammatory response [98]. NF-κB-dependent gene expression requires the involvement of histone acetylase, which will decompress the repressed chromatin environment through histone acetylation [99][100]. Several inflammatory lung diseases are associated with increased H3 acetylation in a specific promoter region that is regulated by NF-κB. Histone modifications at specific acetylation sites (H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K36ac, H2B1KK120ac, H2B2BK20ac, H2BK16ac, H2BK20ac, H2BK108ac, H2BK116ac, and H2BK120ac upregulation; H2BK5, H2BK11 downregulation) are involved in the pathogenesis of asthma [101]. CBP/p300 acetyltransferase promotes the transcription of various proinflammatory cytokines, such as IL-1, IL-2, IL-8, and IL-12, by acetylating histones associated with the promoter region of these genes [98]. The enhanced expression of TNF-α upon LPS stimulation is associated with epigenetic remodeling of the TNF-α locus, which is characterized by increased histone 4 acetylation [102]. In paraquat-induced pulmonary fibrosis, elevated IL-6 expression depends on acetylation of H3K9ac in the IL6 promoter region [103].
4.3. Epigenetic Modulation of the Immune Function
5. Nutrition, Inflammation, and Epigenetics
5.1. Role of Nutrition in Inflammation
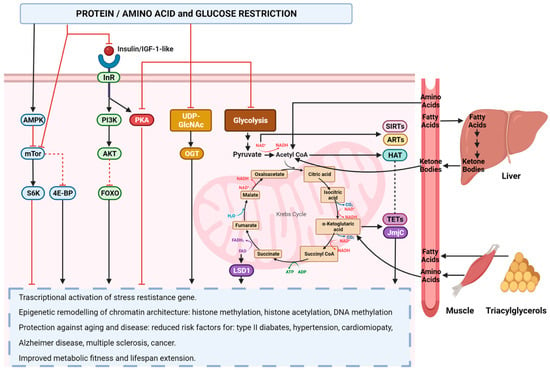
5.2. Role of Nutrition in Epigenetic Reprogramming
6. Inflammation, Epigenetics, and Cancer
6.1. Inflammation in Tumor Onset and Progression
6.2. Epigenetic Alterations in Cancer
6.3. Inflammation-Induced Epigenetic Alterations in Tumor Immune Microenvironment
6.4. Nutrition and Cancer
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472.
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89.
- Cortellino, S.; Longo, V.D. Metabolites and Immune Response in Tumor Microenvironments. Cancers 2023, 15, 3898.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Martínez-Garay, C.; Djouder, N. Dietary interventions and precision nutrition in cancer therapy. Trends Mol. Med. 2023, 29, 489–511.
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218.
- Balka, K.R.; De Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869.
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737.
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169.
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072.
- Imaizumi, T.; Aratani, S.; Nakajima, T.; Carlson, M.; Matsumiya, T.; Tanji, K.; Ookawa, K.; Yoshida, H.; Tsuchida, S.; McIntyre, T.M.; et al. Retinoic acid-inducible gene-I is induced in endothelial cells by LPS and regulates expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279.
- Imaizumi, T.; Hatakeyama, M.; Yamashita, K.; Yoshida, H.; Ishikawa, A.; Taima, K.; Satoh, K.; Mori, F.; Wakabayashi, K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothelium 2004, 11, 169–173.
- Imaizumi, T.; Matsumiya, T.; Yoshida, H.; Naraoka, T.; Uesato, R.; Ishibashi, Y.; Ota, K.; Toh, S.; Fukuda, S.; Satoh, K. Tumor-necrosis factor-alpha induces retinoic acid-inducible gene-I in rheumatoid fibroblast-like synoviocytes. Immunol. Lett. 2009, 122, 89–93.
- Sakaki, H.; Imaizumi, T.; Matsumiya, T.; Kusumi, A.; Nakagawa, H.; Kubota, K.; Nishi, N.; Nakamura, T.; Hirashima, M.; Satoh, K.; et al. Retinoic acid-inducible gene-I is induced by interleukin-1beta in cultured human gingival fibroblasts. Oral. Microbiol. Immunol. 2005, 20, 47–50.
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657.
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791.
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630.
- Wu, J.; Dobbs, N.; Yang, K.; Yan, N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020, 53, 115–126.e5.
- Gazi, U.; Martinez-Pomares, L. Influence of the mannose receptor in host immune responses. Immunobiology 2009, 214, 554–561.
- Miller, J.L.; de Wet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17.
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van Het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 2007, 26, 605–616.
- Hodges, A.; Sharrocks, K.; Edelmann, M.; Baban, D.; Moris, A.; Schwartz, O.; Drakesmith, H.; Davies, K.; Kessler, B.; McMichael, A.; et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat. Immunol. 2007, 8, 569–577.
- Rothfuchs, A.G.; Bafica, A.; Feng, C.G.; Egen, J.G.; Williams, D.L.; Brown, G.D.; Sher, A. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J. Immunol. 2007, 179, 3463–3471.
- Van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601.
- Richard, M.; Thibault, N.; Veilleux, P.; Gareau-Pagé, G.; Beaulieu, A.D. Granulocyte macrophage-colony stimulating factor reduces the affinity of SHP-2 for the ITIM of CLECSF6 in neutrophils: A new mechanism of action for SHP-2. Mol. Immunol. 2006, 43, 1716–1721.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287.
- Inohara, N.; Koseki, T.; Lin, J.; del Peso, L.; Lucas, P.C.; Chen, F.F.; Ogura, Y.; Núñez, G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J. Biol. Chem. 2000, 275, 27823–27831.
- Abbott, D.W.; Wilkins, A.; Asara, J.M.; Cantley, L.C. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004, 14, 2217–2227.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222.
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206.
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298.
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314.
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960.
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863.
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 2011, 187, 4835–4843.
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27.
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189.
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022.
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495.
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343.
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299.
- Van Vugt, M.A.; Parkes, E.E. When breaks get hot: Inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 2022, 8, 174–189.
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132.
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465.
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes. Dev. 2017, 31, 353–369.
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612.
- Hinz, M.; Stilmann, M.; Arslan, S.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74.
- Fang, L.; Choudhary, S.; Zhao, Y.; Edeh, C.B.; Yang, C.; Boldogh, I.; Brasier, A.R. ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014, 42, 8416–8432.
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717.
- Coquel, F.; Silva, M.J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61.
- Gratia, M.; Rodero, M.P.; Conrad, C.; Bou Samra, E.; Maurin, M.; Rice, G.I.; Duffy, D.; Revy, P.; Petit, F.; Dale, R.C.; et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 2019, 216, 1199–1213.
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199.
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100.
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679.
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes. Dev. 2011, 25, 350–362.
- Zhao, S.; Goewey Ruiz, J.A.; Sebastian, M.; Kidane, D. Defective DNA polymerase beta invoke a cytosolic DNA mediated inflammatory response. Front. Immunol. 2022, 13, 1039009.
- Yamaguchi, K.; Kajikawa, M.; Okada, N. LINE retrotransposition and host DNA repair machinery. Mob. Genet. Elem. 2015, 5, 92–97.
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921.
- Kazazian, H.H.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370.
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyö, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596, 43–53.
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178.
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805.
- Goulielmaki, E.; Tsekrekou, M.; Batsiotos, N.; Ascensão-Ferreira, M.; Ledaki, E.; Stratigi, K.; Chatzinikolaou, G.; Topalis, P.; Kosteas, T.; Altmüller, J.; et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Commun. 2021, 12, 3153.
- Chang, E.Y.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol. 2017, 216, 3991–4005.
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 2019, 47, 3485–3502.
- Barnhoorn, S.; Uittenboogaard, L.M.; Jaarsma, D.; Vermeij, W.P.; Tresini, M.; Weymaere, M.; Menoni, H.; Brandt, R.M.; de Waard, M.C.; Botter, S.M.; et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 2014, 10, e1004686.
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365.
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647.
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309.
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160.
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Anti-tumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852.
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470.
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Anti-tumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5.
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8. Cancer Discov. 2019, 9, 722–737.
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143.
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853.
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481.
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16.
- Cheung, P.; Lau, P. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 2005, 19, 563–573.
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318.
- Narlikar, G.J.; Fan, H.Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002, 108, 475–487.
- Youn, H.D. Methylation and demethylation of DNA and histones in chromatin: The most complicated epigenetic marker. Exp. Mol. Med. 2017, 49, e321.
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034.
- Shi, Y.G.; Tsukada, Y. The discovery of histone demethylases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017947.
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356.
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372.
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated with Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346.
- Boyes, J.; Bird, A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 1991, 64, 1123–1134.
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312.
- Villagra, A.; Sotomayor, E.M.; Seto, E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2010, 29, 157–173.
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Calao, M.; Burny, A.; Quivy, V.; Dekoninck, A.; Van Lint, C. A pervasive role of histone acetyltransferases and deacetylases in an NF-kappaB-signaling code. Trends Biochem. Sci. 2008, 33, 339–349.
- Ren, Y.; Li, M.; Bai, S.; Kong, L.; Su, X. Identification of histone acetylation in a murine model of allergic asthma by proteomic analysis. Exp. Biol. Med. 2021, 246, 929–939.
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 2012, 48, 28–38.
- Hu, L.; Yu, Y.; Huang, H.; Fan, H.; Yin, C.; Li, K.; Fulton, D.J.; Chen, F. Epigenetic Regulation of Interleukin 6 by Histone Acetylation in Macrophages and Its Role in Paraquat-Induced Pulmonary Fibrosis. Front. Immunol. 2016, 7, 696.
- Sadler, A.J.; Suliman, B.A.; Yu, L.; Yuan, X.; Wang, D.; Irving, A.T.; Sarvestani, S.T.; Banerjee, A.; Mansell, A.S.; Liu, J.P.; et al. The acetyltransferase HAT1 moderates the NF-κB response by regulating the transcription factor PLZF. Nat. Commun. 2015, 6, 6795.
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953.
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245.
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387.
- Balada, E.; Castro-Marrero, J.; Felip, L.; Ordi-Ros, J.; Vilardell-Tarrés, M. Associations between the expression of epigenetically regulated genes and the expression of DNMTs and MBDs in systemic lupus erythematosus. PLoS ONE 2012, 7, e45897.
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.J.; et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922.
- Karatzas, P.S.; Mantzaris, G.J.; Safioleas, M.; Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine 2014, 93, e309.
- Akbaba, T.H.; Akkaya-Ulum, Y.Z.; Tavukcuoglu, Z.; Bilginer, Y.; Ozen, S.; Balci-Peynircioglu, B. Inflammation-related differentially expressed common miRNAs in systemic autoinflammatory disorders patients can regulate the clinical course. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S132), 109–117.
- Hussain, N.; Zhu, W.; Jiang, C.; Xu, J.; Wu, X.; Geng, M.; Hussain, S.; Cai, Y.; Xu, K.; Xu, P.; et al. Down-regulation of miR-10a-5p in synoviocytes contributes to TBX5-controlled joint inflammation. J. Cell Mol. Med. 2018, 22, 241–250.
- Aznaourova, M.; Janga, H.; Sefried, S.; Kaufmann, A.; Dorna, J.; Volkers, S.M.; Georg, P.; Lechner, M.; Hoppe, J.; Dökel, S.; et al. Noncoding RNA. Proc. Natl. Acad. Sci. USA 2020, 117, 9042–9053.
- Kersh, E.N.; Fitzpatrick, D.R.; Murali-Krishna, K.; Shires, J.; Speck, S.H.; Boss, J.M.; Ahmed, R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 2006, 176, 4083–4093.
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763.
- Chang, S.; Collins, P.L.; Aune, T.M. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J. Immunol. 2008, 181, 8372–8381.
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832.
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.A.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429.
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315.
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573.
- Huang, D.; Chen, J.; Yang, L.; Ouyang, Q.; Li, J.; Lao, L.; Zhao, J.; Liu, J.; Lu, Y.; Xing, Y.; et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat. Immunol. 2018, 19, 1112–1125.
- Pyfrom, S.C.; Quinn, C.C.; Dorando, H.K.; Luo, H.; Payton, J.E. BCALM (AC099524.1) Is a Human B Lymphocyte-Specific Long Noncoding RNA That Modulates B Cell Receptor-Mediated Calcium Signaling. J. Immunol. 2020, 205, 595–607.
- Li, J.; Tian, J.; Lu, J.; Wang, Z.; Ling, J.; Wu, X.; Yang, F.; Xia, Y. LncRNA GAS5 inhibits Th17 differentiation and alleviates immune thrombocytopenia via promoting the ubiquitination of STAT3. Int. Immunopharmacol. 2020, 80, 106127.
- Fu, J.; Shi, H.; Wang, B.; Zhan, T.; Shao, Y.; Ye, L.; Wu, S.; Yu, C.; Zheng, L. LncRNA PVT1 links Myc to glycolytic metabolism upon CD4. J. Autoimmun. 2020, 107, 102358.
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101.
- Sears, B.; Lawren, B. The Zone: A Dietary Road Map; ReganBooks: New York, NY, USA, 1995; p. xvii. 328p.
- Brenner, R.R. Hormonal modulation of delta6 and delta5 desaturases: Case of diabetes. Prostaglandins Leukot. Essent. Fatty Acids 2003, 68, 151–162.
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell Biochem. 2018, 119, 105–110.
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669.
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400.
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245.
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560.
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774.
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135.
- McArthur, S.; Juban, G.; Gobbetti, T.; Desgeorges, T.; Theret, M.; Gondin, J.; Toller-Kawahisa, J.E.; Reutelingsperger, C.P.; Chazaud, B.; Perretti, M.; et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J. Clin. Investig. 2020, 130, 1156–1167.
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90.
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575.
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823.
- Cortellino, S.; Raveane, A.; Chiodoni, C.; Delfanti, G.; Pisati, F.; Spagnolo, V.; Visco, E.; Fragale, G.; Ferrante, F.; Magni, S.; et al. Fasting renders immunotherapy effective against low-immunogenic breast cancer while reducing side effects. Cell Rep. 2022, 40, 111256.
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719.
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711.
- Keating, S.T.; El-Osta, A. Epigenetics and metabolism. Circ. Res. 2015, 116, 715–736.
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41.
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282.
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5.
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673.
- Drutman, S.B.; Haerynck, F.; Zhong, F.L.; Hum, D.; Hernandez, N.J.; Belkaya, S.; Rapaport, F.; de Jong, S.J.; Creytens, D.; Tavernier, S.J.; et al. Homozygous. Proc. Natl. Acad. Sci. USA 2019, 116, 19055–19063.
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17.
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment. Cell Melanoma Res. 2012, 25, 506–513.
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.L.; Fock, K.M.; Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: A case-control study and gene expression analyses. PLoS ONE 2015, 10, e0117870.
- Miskiewicz, A.; Szparecki, G.; Durlik, M.; Rydzewska, G.; Ziobrowski, I.; Górska, R. The Q705K and F359L Single-Nucleotide Polymorphisms of NOD-Like Receptor Signaling Pathway: Association with Chronic Pancreatitis, Pancreatic Cancer, and Periodontitis. Arch. Immunol. Ther. Exp. 2015, 63, 485–494.
- Zhao, X.; Zhang, C.; Hua, M.; Wang, R.; Zhong, C.; Yu, J.; Han, F.; He, N.; Zhao, Y.; Liu, G.; et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 2017, 8, 108571–108583.
- Chai, D.; Zhang, Z.; Shi, S.Y.; Qiu, D.; Zhang, C.; Wang, G.; Fang, L.; Li, H.; Tian, H.; Zheng, J. Absent in melanoma 2-mediating M1 macrophages facilitate tumor rejection in renal carcinoma. Transl. Oncol. 2021, 14, 101018.
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586.
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101.
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375.
- Huang, Q.; Lan, F.; Wang, X.; Yu, Y.; Ouyang, X.; Zheng, F.; Han, J.; Lin, Y.; Xie, Y.; Xie, F.; et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 2014, 13, 18.
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277.
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155.
- Jin, H.; Ko, Y.S.; Kim, H.J. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int. J. Oncol. 2018, 53, 1953–1966.
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499.
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic Plasticity and Cell Fate Decisions in Cancer: Insights from Dynamical Systems Theory. Cancers 2017, 9, 70.
- Sacchetti, A.; Teeuwssen, M.; Verhagen, M.; Joosten, R.; Xu, T.; Stabile, R.; van der Steen, B.; Watson, M.M.; Gusinac, A.; Kim, W.K.; et al. Phenotypic plasticity underlies local invasion and distant metastasis in colon cancer. eLife 2021, 10, e61461.
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80.
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat. Rev. Cancer 2021, 21, 413–430.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231.
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618.
- Avvakumov, N.; Côté, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407.
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231.
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18.
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18.
- Modena, P.; Lualdi, E.; Facchinetti, F.; Galli, L.; Teixeira, M.R.; Pilotti, S.; Sozzi, G. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005, 65, 4012–4019.
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349.
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178.
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520.
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065.
- Ding, J.; Zhang, Z.M.; Xia, Y.; Liao, G.Q.; Pan, Y.; Liu, S.; Zhang, Y.; Yan, Z.S. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br. J. Cancer 2013, 109, 994–1003.
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347.
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477.
- De Koning, L.; Savignoni, A.; Boumendil, C.; Rehman, H.; Asselain, B.; Sastre-Garau, X.; Almouzni, G. Heterochromatin protein 1alpha: A hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1, 178–191.
- Coles, A.H.; Jones, S.N. The ING gene family in the regulation of cell growth and tumorigenesis. J. Cell Physiol. 2009, 218, 45–57.
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97.
- Tricarico, R.; Cortellino, S.; Riccio, A.; Jagmohan-Changur, S.; Van der Klift, H.; Wijnen, J.; Turner, D.; Ventura, A.; Rovella, V.; Percesepe, A.; et al. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget 2015, 6, 42892–42904.
- Riccio, A.; Aaltonen, L.A.; Godwin, A.K.; Loukola, A.; Percesepe, A.; Salovaara, R.; Masciullo, V.; Genuardi, M.; Paravatou-Petsotas, M.; Bassi, D.E.; et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 1999, 23, 266–268.
- Nejati-Koshki, K.; Roberts, C.T.; Babaei, G.; Rastegar, M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers 2023, 15, 2683.
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380.
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492.
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486.
- Stoitzner, P.; Green, L.K.; Jung, J.Y.; Price, K.M.; Atarea, H.; Kivell, B.; Ronchese, F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008, 57, 1665–1673.
- Zhou, Z.; Chen, H.; Xie, R.; Wang, H.; Li, S.; Xu, Q.; Xu, N.; Cheng, Q.; Qian, Y.; Huang, R.; et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol. 2019, 13, 873–893.
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952.
- Rosenzweig, J.M.; Glenn, J.D.; Calabresi, P.A.; Whartenby, K.A. KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J. Biol. Chem. 2013, 288, 23868–23874.
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223.
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254.
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589.
- Cheng, F.; Lienlaf, M.; Perez-Villarroel, P.; Wang, H.W.; Lee, C.; Woan, K.; Woods, D.; Knox, T.; Bergman, J.; Pinilla-Ibarz, J.; et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60, 44–53.
- Zhao, N.H.; Qian, Y.; Wu, C.S.; Wang, J.W.; Fang, Y.; Fan, X.P.; Gao, S.; Fan, Y.C.; Wang, K. Diagnostic value of NKG2D promoter methylation in hepatitis B virus-associated hepatocellular carcinoma. Biomark. Med. 2019, 13, 1093–1105.
- Stephen, T.L.; Payne, K.K.; Chaurio, R.A.; Allegrezza, M.J.; Zhu, H.; Perez-Sanz, J.; Perales-Puchalt, A.; Nguyen, J.M.; Vara-Ailor, A.E.; Eruslanov, E.B.; et al. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity 2017, 46, 51–64.
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019, 21, 2.
- Key, T.J.; Allen, N.E.; Spencer, E.A.; Travis, R.C. The effect of diet on risk of cancer. Lancet 2002, 360, 861–868.
- Newmark, H.L.; Yang, K.; Kurihara, N.; Fan, K.; Augenlicht, L.H.; Lipkin, M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: A preclinical model for human sporadic colon cancer. Carcinogenesis 2009, 30, 88–92.
- Trichopoulou, A.; Bamia, C.; Lagiou, P.; Trichopoulos, D. Conformity to traditional Mediterranean diet and breast cancer risk in the Greek EPIC (European Prospective Investigation into Cancer and Nutrition) cohort. Am. J. Clin. Nutr. 2010, 92, 620–625.
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62.
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706.
- Ames, B.N. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat. Res. 2001, 475, 7–20.
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276.
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672.
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285.
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 2014, 53, 710–725.
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478.
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948.
- Mocellin, S.; Briarava, M.; Pilati, P. Vitamin B6 and Cancer Risk: A Field Synopsis and Meta-Analysis. J. Natl. Cancer Inst. 2017, 109, djw230.
- Dou, R.; Ng, K.; Giovannucci, E.L.; Manson, J.E.; Qian, Z.R.; Ogino, S. Vitamin D and colorectal cancer: Molecular, epidemiological and clinical evidence. Br. J. Nutr. 2016, 115, 1643–1660.
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Koo, J.; Hood, N. Prognostic effects of 25-hydroxyvitamin D levels in early breast cancer. J. Clin. Oncol. 2009, 27, 3757–3763.
- Tretli, S.; Hernes, E.; Berg, J.P.; Hestvik, U.E.; Robsahm, T.E. Association between serum 25(OH)D and death from prostate cancer. Br. J. Cancer 2009, 100, 450–454.

