Inflammation is a key contributor to both the initiation and progression of tumors, and it can be triggered by genetic instability within tumors, as well as by lifestyle and dietary factors. The inflammatory response plays a critical role in the genetic and epigenetic reprogramming of tumor cells, as well as in the cells that comprise the tumor microenvironment. Cells in the microenvironment acquire a phenotype that promotes immune evasion, progression, and metastasis.
- inflammation
- epigenetic
- DNA repair
- nutrition
- cancer
1. Introduction
2. Inflammation
Inflammation is a highly intricate process that is regulated by the immune system in response to external or internal stimuli that threaten tissue integrity [7]. The innate immune system is primarily responsible for triggering this response. It recognizes molecules or portions of molecules that are released upon cellular stress or tissue injury, such as damage-associated molecular patterns (DAMPs), or are specific to pathogens, such as pathogen-associated molecular patterns (PAMPs). PAMPs and DAMPs bind to pattern recognition receptors (PRRs) located on the cytoplasmic or endosome membrane, activating intracellular signaling cascades that result in the expression of proinflammatory factors [8]. PRRs include toll-like receptors (TLRs), retinoic acid-inducible gene 1-(RIG1)-like receptors (RLRs), cytosolic DNA sensor cyclic GMP–AMP synthase (cGAS) stimulator of interferon genes (STINGs), C-type lectin receptors (CLRs), and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (Figure 1).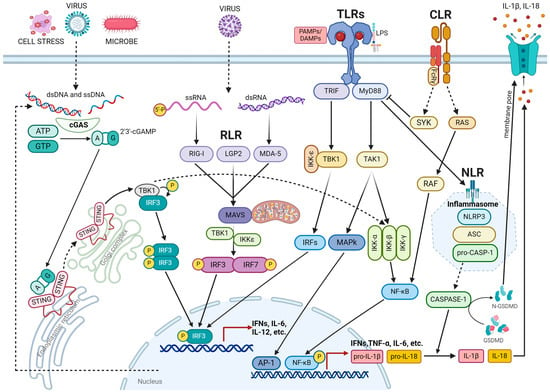
3. DNA Damage Response in Inflammation
3.1. Mechanism Underlying DNA Damage-Induced Inflammation
DNA repair mechanisms and signaling pathways are closely linked to the inflammatory response. DNA damage activates the cGAS-STING pathway, which induces the expression of type I interferons and inflammatory factors. cGAS triggers this process by detecting endogenous DNA released from the nucleus, mitochondria, or micronuclei into the cytoplasm as well as potentially exogenous DNA derived from pathogenic microorganisms [48][49][50][49,50,51]. Micronuclei, which are small, membrane-enclosed structures composed of acentric chromosomal fragments, are generated as a result of defects in DNA repair processes, chromosomal segregation, and non-disjunction. These defects can be caused by mutations in genes involved in the DNA damage response (DDR) and the regulation of the mitotic spindle, or they can be induced by DNA-damaging treatments [51][52][53][52,53,54]. Unrepaired or misrepaired DNA double-strand breaks (DSBs) lead to the formation of acentric chromatids that segregate improperly during anaphase, are excluded from the nucleus, and are enveloped by the nuclear membrane [51][52]. Although the c-GAS-STING pathway serves as the primary mechanism to induce inflammatory responses to genotoxic stress, DDR proteins can also trigger inflammation by directly or indirectly activating NF-kB. Damaged DNA, induced by genotoxic agents, recruits and activates ATM and ATM and RAD3 related (ATR), which can stimulate NF-kB activation through the following: (1) stabilization of GATA4 through inhibition of p62 and autophagic degradation of GATA4 [54][57]; (2) assembly of an alternative STING signaling complex including the tumor suppressor p53 and the E3 ubiquitin ligase TRAF6 [55][58]; (3) degradation of IκBα through the formation of the IκBα-β-TrCP-ubiquitin ligase complex and phosphorylation of RELA (also known as p65) through interaction with protein kinase A (PKA) [56][59].3.2. DNA Repair Deficiency Disorder and Inflammation
The DDR is a vital mechanism that ensures the preservation of genomic integrity and prevents tumor formation by employing an intricate signaling network that detects, signals, and repairs DNA damage. Numerous recent investigations have pointed out that the deletion or mutation of DDR genes, both inherited and spontaneous, as well as the genotoxic stress induced by DNA-damaging treatments, can trigger inflammation by activating the cGAS-STING pathway and other associated signaling cascades [48][57][58][59][60][61][49,62,63,64,65,66]. The deficiency of BRCA1 or BRCA2 tumor suppressors, which play a crucial role in repairing DNA breaks through the promotion of the homologous recombination (HR) DNA repair pathway, and in maintaining the stability of newly synthesized DNA strands by safeguarding stalled replication forks from degradation, also triggers type I IFN signaling and anti-tumor immunity. Cells lacking BRCA1 or BRCA2 exhibit elevated levels of cytosolic DNA and the constitutively active viral response cGAS/STING/TBK1/IRF3 pathway [60][61][65,66]. The c-GAS-STING pathway can also be activated by other proteins involved in DNA repair. For instance, the inactivation of BLM, a DNA helicase involved in the repair of DNA double-strand breaks through the homologous recombination (HR) pathway, can compromise the integrity of DNA by blocking the restart of stalled replication forks [62][69], as well as DNA double-strand break resection [63][70] and Holliday junction dissolution. BLM mutations are responsible for Bloom syndrome, a genetic disorder characterized by genetic instability and an increased risk of cancer. Even the inhibition of proteins involved in base excision repair (BER) can elicit an interferon response through activation of the cGAS pathway. Mutations in the DNA polymerase beta (POLB) protein, which is involved in BER and fills in single-nucleotide gaps, result in replication-associated genomic instability and inflammation-associated carcinogenesis in mice. This occurs because failure to repair the damage leads to the formation of single-strand breaks (SSBs), which are converted into double-strand breaks (DSBs) during the S-phase of DNA replication. The accumulation of DSBs causes mitotic dysfunction, leading to an increase in micronuclei formation due to the mis-segregation of broken chromosomes during mitosis. This, in turn, triggers a c-GAS-mediated inflammatory response by releasing cytosolic DNA [64][72].3.3. Inflammation Promoted by Transposable Element (TE) Instability
Dysfunctions in the DNA damage response pathway can result in a strong inflammatory response by accelerating the activation of transposable elements (TEs), which are DNA sequences capable of self-replication and relocating within the same genome [65][73]. The eukaryotic genome is primarily composed of repetitive DNA sequences, including satellite DNA and TEs. TEs are divided into two groups: retrotransposable elements (RTEs), such as long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), and endogenous retroviruses (ERVs) [66][67][68][74,75,76]. Active RTEs produce cytosolic DNA (cDNAs) that activate the cGAS-STING pathway and initiate the production of pro-inflammatory cytokines, such as IFNα, IFNβ, IL-6, and TNF, through NF-κB and IRF3 stimulation. Although the mechanism underlying RTE-induced inflammation remains unclear, it is believed that abnormal DNA damage repair mechanisms can result in the accumulation and dispersion of cDNA in the cytoplasm by affecting the integration of RTE DNA into the genome.3.4. Inflammation Triggered by R-Loops Resolving Defects
Inflammation can arise from disruptions in the mechanisms responsible for maintaining and resolving R-loops. R-loops are nucleic acid structures comprising DNA-RNA hybrids and displaced single-stranded DNA resulting from transcription and replication [69][78]. If these structures persist for an extended period, endonucleases cut the exposed single-stranded DNA, leading to single- and double-strand breaks. Alterations in helicases, such as SENATAXIN, BLM, and WRN, and endonucleases, such as ERCC1 and XPG, can impair R-loop resolution [70][71][72][73][74][79,80,81,82,83]. The loss of tumor suppressors BRCA1 and BRCA2, which are involved in repairing double-strand breaks, can contribute to an abnormal accumulation of R-loops and cytosolic DNA, resulting in a heightened inflammatory response [75][76][84,85].3.5. Anti-Tumor Cytotoxicity Mediated by Inflammatory Response
The effectiveness of chemotherapeutic agents, including anthracyclines, oxaliplatin, and doxorubicin, depends, in part, on their ability to induce anti-tumor immune responses through type I IFN signaling and ISGs [77][78][86,87]. Similarly, the anti-tumor immunity triggered by ionizing radiation (IR) is dependent on the cGAS-STING pathway [79][88]. Recent studies have identified the accumulation of cytosolic DNA following chemotherapy or radiation as the trigger for cGAS-STING activation, leading to the induction of IFN and ISGs [52][53][80][53,54,89]. Cytosolic DNA is generated as a result of double-strand break repair processes triggered by ionizing radiation or chemotherapy. Specifically, enzymes such as BLM helicase and exonuclease 1 (EXO1), which play a crucial role in the final resection of DNA during double-strand break repair, generate these DNA fragments that can either be enclosed in micronuclei or migrate to the cytoplasm, where they activate the cGAS-STING pathway [53][80][54,89]. Inhibiting or deactivating DDR pathways can enhance the inflammatory response and improve the immune response induced by genotoxic stress agents. PARP inhibitors (PARPi) increase endogenous genomic instability in BRCA1/2-deficient cells and tumors by blocking DNA repair mechanisms and replication fork progression, which leads to an increase in cytosolic DNA and activates the cGAS-STING pathway [81][82][83][91,92,93]. As a result, ISGs are expressed in BRCA1/2-deficient tumor cells and stimulate T-cell infiltration and activation, ultimately leading to tumor eradication in BRCA1/2-deficient mouse models of ovarian and breast cancer [84][94].4. Epigenetics and Inflammation
4.1. Epigenetic Regulatory Mechanisms
Epigenetic modulators are divided into (1) writers, which add covalent modifications to histones or DNA; (2) erasers, which remove histones and DNA modifications; and (3) readers, which recognize and bind epigenetic modifications [85][86][87][96,97,98] (Figure 2).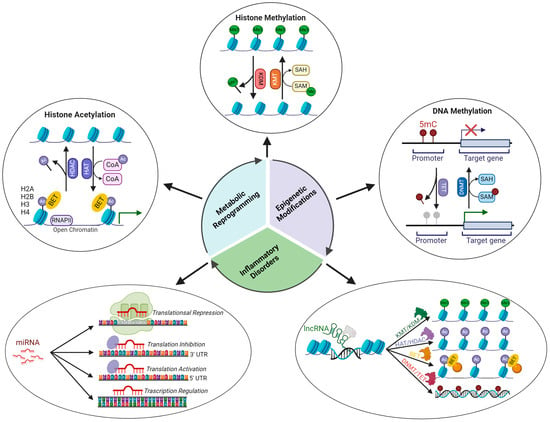
4.2. Epigenetic Signatures Underlying Inflammation
Histone acetylation. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate the expression of several genes involved in the inflammatory response [98][130]. NF-κB-dependent gene expression requires the involvement of histone acetylase, which will decompress the repressed chromatin environment through histone acetylation [99][100][131,132]. Several inflammatory lung diseases are associated with increased H3 acetylation in a specific promoter region that is regulated by NF-κB. Histone modifications at specific acetylation sites (H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K36ac, H2B1KK120ac, H2B2BK20ac, H2BK16ac, H2BK20ac, H2BK108ac, H2BK116ac, and H2BK120ac upregulation; H2BK5, H2BK11 downregulation) are involved in the pathogenesis of asthma [101][133]. CBP/p300 acetyltransferase promotes the transcription of various proinflammatory cytokines, such as IL-1, IL-2, IL-8, and IL-12, by acetylating histones associated with the promoter region of these genes [98][130]. The enhanced expression of TNF-α upon LPS stimulation is associated with epigenetic remodeling of the TNF-α locus, which is characterized by increased histone 4 acetylation [102][134]. In paraquat-induced pulmonary fibrosis, elevated IL-6 expression depends on acetylation of H3K9ac in the IL6 promoter region [103][135].
4.3. Epigenetic Modulation of the Immune Function
In addition to their role in the inflammatory response, epigenetic modifications are crucial in the differentiation and maintenance of T-lymphocyte functions. In the inactive or naive state, DNMT1 methylates the promoter regions of transcription factors that define the T-cell lineage, thereby suppressing their expression. Upon antigenic stimulation, TET proteins assist in removing the methyl group from gene promoters, triggering the transcription of specific transcription factors, such as T-bet, GATA-3, RORgt, and Foxp3, which are expressed in each T-cell subset [114][115][160,161]. The differentiation of T lymphocytes into specific subsets, such as Th1 and TH2, involves epigenetic modifications. For instance, helper T-lymphocyte polarization into the Th1 subset is characterized by an increase in H4 acetylation on the Ifn-γ promoter [116][162], while polarization into the Th2 subset results in the repression of Ifn-γ via H3K27me3 methylation by EZH2 [117][163]. The differentiation of CD8 T cells into effector T cells, such as cytotoxic T cells, is accompanied by increased expression of IFN-γ, perforin (PRF), and granzyme B (GZB) genes due to demethylation of their promoter regions [118][119][164,165]. LncRNAs also play a significant role in the activation and differentiation of lymphocytes. Several lncRNAs, such as NRON, NKILA, BCALM, GAS5, and PVT1, regulate the activation of T lymphocytes and the TCR/BCR signaling pathway by modulating the activity of NFAT, NFκB, and MYC [120][121][122][123][124][166,167,168,169,170].5. Nutrition, Inflammation, and Epigenetics
5.1. Role of Nutrition in Inflammation
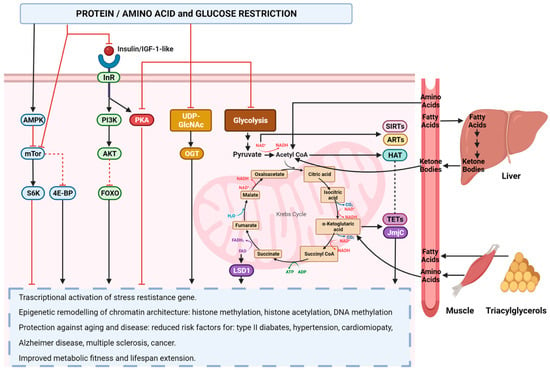
Nutrition exerts a critical influence on the inflammatory response, as it regulates diverse genetic and epigenetic mechanisms involved in this process. The inflammatory response is dependent on the activity of eicosanoid hormones, specialized pro-resolution mediators (SPMs), and modulatory genes [125][173]. Eicosanoids are primarily derived from arachidonic acids, which are membrane phospholipids that are released upon the activation of phospholipase A2 by an inflammatory insult. The intensity of the inflammatory response is proportional to the amount of arachidonic acid present in the membrane [126][174]. The production of arachidonic acid is contingent upon the activity and regulation of two enzymes, delta-6-desaturase and delta-5-desaturase, which are responsible for converting linoleic acid to arachidonic acid. Elevated levels of insulin activate these enzymes, and insulin resistance induced by cytokines, such as TNF-α, can contribute to the production of arachidonic acid [127][128][175,176]. Specialized pro-resolving mediators (SPMs) are hormones derived from EPA, DHA, and docosapentaenoic acid (DPA), which play a crucial role in the resolution of residual inflammation [129][180]. These SPMs include resolvins, maresins, and protectins and exert their anti-inflammatory effects by inhibiting the migration of neutrophils to the site of injury, promoting the transition of macrophages from the pro-inflammatory M1 subtype to the anti-inflammatory M2 subtype and facilitating the removal of apoptotic cells by professional and non-professional phagocytes [130][181]. The 5′-adenosine monophosphate-activated protein kinase (AMPK) gene is the most critical regulator of inflammation modulation, as it inhibits NF-kB. AMPK regulates cellular energy homeostasis and is activated in response to energy stress, leading to an increase in the AMP/ATP ratio. Activation of AMPK facilitates the metabolic shift from anabolism to catabolism, initiates autophagy, which is vital for supplying energy, metabolites, and biosynthetic intermediates necessary for healing inflammatory lesions, and stimulates mitophagy to replace damaged mitochondria with functional organelles capable of providing the energy required for resolving inflammatory damage [131][132][133][134][183,184,185,186]. AMPK activity is positively regulated by the SPM signaling pathway, as the binding of SPM, such as Resolvin D1 (RvD1), to SPM receptors, such as FPR2/ALX, promotes AMPK activation [135][187]. Polyphenols, natural compounds derived from plants, fruits, and vegetables with antioxidant properties, can reduce the inflammatory response and promote resolution of the inflammatory state through the indirect activation of AMPK. Polyphenols, particularly anthocyanins, have been demonstrated to exert a positive regulatory effect on AMPK through the activation of sirtuins. Sirtuins, in turn, stimulate the transcription of hepatic kinase B1 (LKB1), which subsequently promotes the transcription of AMPK by deacetylating the AMPK promoter [136][137][188,189]. However, an essential requirement for an effective anti-inflammatory diet is a low-calorie intake. Calorie restriction (CR) exerts potent anti-inflammatory effects through the activation of the anti-stress response, which leads to a reduction in oxidative stress and the protection of DNA from oxidative damage by activating AMPK and suppressing protein kinase A (PKA) and mammalian target of rapamycin (mTOR) signaling pathways, which are triggered by glucose and amino acids, such as threonine and valine [138][191] (Figure 3).