 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shuanglin Qin | -- | 4225 | 2024-03-05 08:22:26 | | | |
| 2 | Mona Zou | Meta information modification | 4225 | 2024-03-06 09:37:48 | | |
Video Upload Options
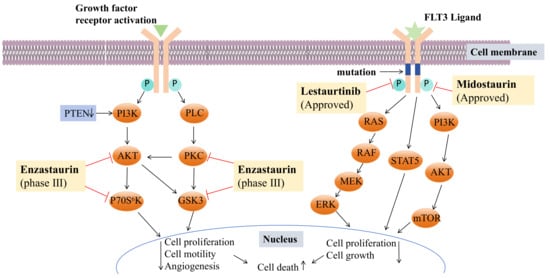
Indole is a multifunctional active pharmacophore and a heterocyclic compound widely present in natural and synthetic compounds with biological activity. Indole alkaloids from natural sources display diverse mechanisms and structures and exert anticancer potential through various antiproliferation mechanisms. Thus, indole alkaloids play a significant role in the discovery of new anticancer drugs. Scientists have subsequently isolated various new bisindole alkaloids from marine organisms, especially deep-water sponges. They are usually extracted with organic solvents (e.g., methanol, ethyl acetate). And the extracts were concentrated and partitioned between organic and aqueous phases. The organic phase is separated with chromatography separation technology, including silica gel column chromatography, high-performance liquid chromatography, etc. Most marine-derived bisindoles exist in solid form, which makes it convenient for us to determine their absolute configuration by single crystal. A number of marine-derived bisindoles exhibit strong and varied biological activities. Due to their unique biological activities and chemical structures, they have become a research focal point in pharmaceutical chemistry as lead compounds for new drug development. It is noteworthy that many drugs based on marine-derived bisindoles have been approved or are currently in clinical research, such as midostaurin, lestaurtinib, and enzastaurin. These drug molecules have shown potent selective anti-tumor effects.
1. Topsentin Family Bisindole Alkaloids and Their Derivatives
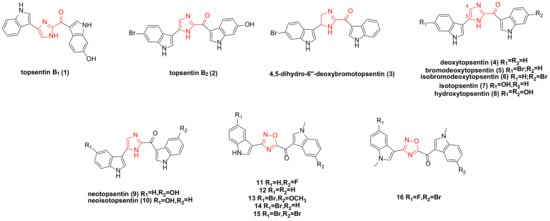
2. Nortopsentin Family Bisindole Alkaloids and Their Derivatives
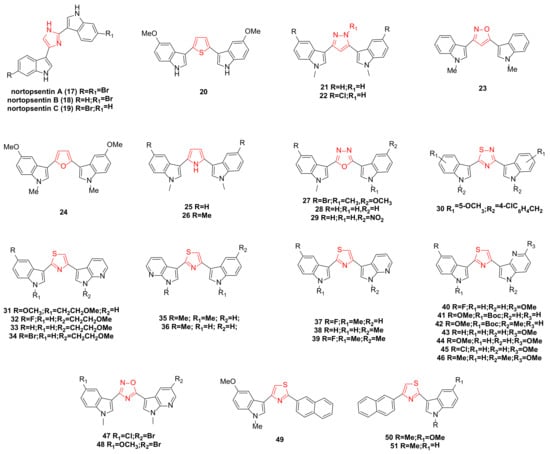
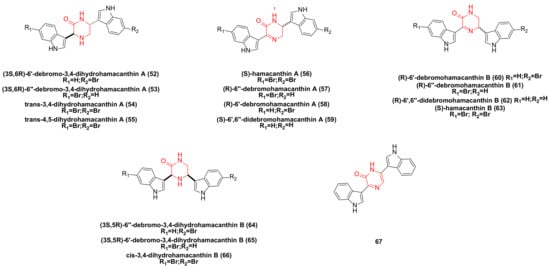
3. Hamacanthin Family Bisindole Alkaloids and Their Derivatives
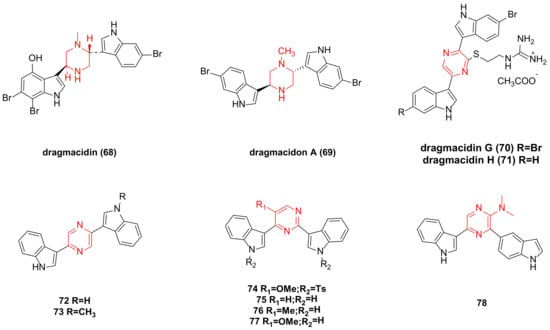
4. Dragmacidin Family Bisindole Alkaloids and Their Derivatives
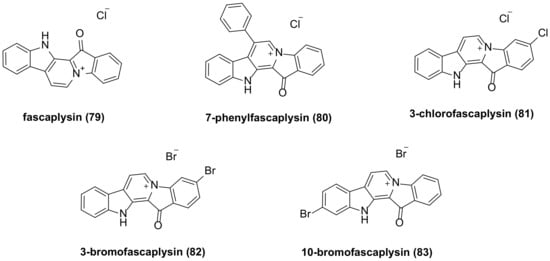
5. Fascaplysin Family Bisindole Alkaloids and Their Derivatives
6. Bisindole Acetylamine Alkaloids and Their Derivatives
7. Acyclic Structure-Linked Bisindole Alkaloids and Their Derivatives
8. Indolecarbazole Bisindole Alkaloids and Their Derivatives
References
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, Bromotopsentin, and Dihydrodeoxybromotopsentin: Antiviral and Antitumor Bis(Indolyl)Imidazoles from Caribbean Deep-Sea Sponges of the Family Halichondriidae. Structural and Synthetic Studies. J. Org. Chem. 1988, 53, 5446–5453.
- Burres, N.S.; Barber, D.A.; Gunasekera, S.P.; Shen, L.L.; Clement, J.J. Antitumor Activity and Biochemical Effects of Topsentin. Biochem. Pharmacol. 1991, 42, 745–751.
- Bartik, K.; Braekman, J.-C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentins, New Toxic Bis-Indole Alkaloids from the Marine Sponge Topsentia Genitrix. Can. J. Chem. 1987, 65, 2118–2121.
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from the Mediterranean Marine Sponge Rhaphisia Lacazei. J. Nat. Prod. 2000, 63, 447–451.
- Hitora, Y.; Takada, K.; Ise, Y.; Okada, S.; Matsunaga, S. Dragmacidins G and H, Bisindole Alkaloids Tethered by a Guanidino Ethylthiopyrazine Moiety, from a Lipastrotethya Sp. Marine Sponge. J. Nat. Prod. 2016, 79, 2973–2976.
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Lee, T.-H.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Antimicrobial Activity and Cytotoxicity of Bis(Indole) Alkaloids from the Sponge Spongosorites sp. Biol. Pharm. Bull. 2006, 29, 570–573.
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and Antifungal Imidazolediylbis from the Sponge Spongosorites Ruetzleri. J. Org. Chem. 1991, 56, 4304–4307.
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.-R.; Sim, C.J. New Bis(Indole) Alkaloids of the Topsentin Class from the Sponge Spongosorites genitrix. J. Nat. Prod. 1999, 62, 647–649.
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Sim, C.J.; Im, K.S.; Jung, J.H. Cytotoxic Bisindole Alkaloids from a Marine Sponge Spongosorites sp. J. Nat. Prod. 2005, 68, 711–715.
- Cascioferro, S.; Li Petri, G.; Parrino, B.; El Hassouni, B.; Carbone, D.; Arizza, V.; Perricone, U.; Padova, A.; Funel, N.; Peters, G.J.; et al. 3-(6-Phenylimidazo Thiadiazol-2-Yl)-1H-Indole Derivatives as New Anticancer Agents in the Treatment of Pancreatic Ductal Adenocarcinoma. Molecules 2020, 25, 329.
- Li Petri, G.; Cascioferro, S.; El Hassouni, B.; Carbone, D.; Parrino, B.; Cirrincione, G.; Peters, G.J.; Diana, P.; Giovannetti, E. Biological Evaluation of the Antiproliferative and Anti-Migratory Activity of a Series of 3-(6-Phenylimidazo Thiadiazol-2-Yl)-1 H -Indole Derivatives Against Pancreatic Cancer Cells. Anticancer Res. 2019, 39, 3615–3620.
- Mérour, J.-Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The Azaindole Framework in the Design of Kinase Inhibitors. Molecules 2014, 19, 19935–19979.
- Parrino, B.; Carbone, A.; Spanò, V.; Montalbano, A.; Giallombardo, D.; Barraja, P.; Attanzio, A.; Tesoriere, L.; Sissi, C.; Palumbo, M.; et al. Aza-Isoindolo and Isoindolo-Azaquinoxaline Derivatives with Antiproliferative Activity. Eur. J. Med. Chem. 2015, 94, 367–377.
- Parrino, B.; Carbone, A.; Ciancimino, C.; Spanò, V.; Montalbano, A.; Barraja, P.; Cirrincione, G.; Diana, P.; Sissi, C.; Palumbo, M.; et al. Water-Soluble Isoindolo Quinoxalin-6-Imines: In Vitro Antiproliferative Activity and Molecular Mechanism(s) of Action. Eur. J. Med. Chem. 2015, 94, 149–162.
- Carbone, D.; Parrino, B.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cirrincione, G.; Diana, P. 1,2,4-Oxadiazole Topsentin Analogs with Antiproliferative Activity against Pancreatic Cancer Cells, Targeting GSK3β Kinase. ChemMedChem 2021, 16, 537–554.
- Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2021, 27, 19.
- Kamel, M.M.; Abdel-hameid, M.K.; El-Nassan, H.B.; El-Khouly, E.A. Recent Advances in the Synthesis and Biological Applications of Nortopsentin Analogs. Chem. Heterocycl. Compd. 2020, 56, 499–502.
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Martorana, A.; Dattolo, G.; Gia, O.; Via, L.D.; Cirrincione, G. Synthesis and Antitumor Properties of 2,5-Bis(3′-Indolyl)Thiophenes: Analogues of Marine Alkaloid Nortopsentin. Bioorg. Med. Chem. Lett. 2007, 17, 2342–2346.
- Diana, P.; Carbone, A.; Barraja, P.; Martorana, A.; Gia, O.; DallaVia, L.; Cirrincione, G. 3,5-Bis(3′-Indolyl)Pyrazoles, Analogues of Marine Alkaloid Nortopsentin: Synthesis and Antitumor Properties. Bioorg. Med. Chem. Lett. 2007, 17, 6134–6137.
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and Antitumor Activity of 2,5-Bis(3′-Indolyl)-Furans and 3,5-Bis(3′-Indolyl)-Isoxazoles, Nortopsentin Analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529.
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.-H. Synthesis and Antiproliferative Activity of 2,5-Bis(3′-Indolyl)Pyrroles, Analogues of the Marine Alkaloid Nortopsentin. Mar. Drugs 2013, 11, 643–654.
- Sreenivasulu, R.; Tej, M.B.; Jadav, S.S.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, Anticancer Evaluation and Molecular Docking Studies of 2,5-Bis(Indolyl)-1,3,4-Oxadiazoles, Nortopsentin Analogues. J. Mol. Struct. 2020, 1208, 127875.
- Kumar, D.; Kumar, N.M.; Chang, K.-H.; Gupta, R.; Shah, K. Synthesis and In-Vitro Anticancer Activity of 3,5-Bis(Indolyl)-1,2,4-Thiadiazoles. Bioorg. Med. Chem. Lett. 2011, 21, 5897–5900.
- Parrino, B.; Attanzio, A.; Spanò, V.; Cascioferro, S.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Diana, P.; Cirrincione, G.; Carbone, A. Synthesis, Antitumor Activity and CDK1 Inhibiton of New Thiazole Nortopsentin Analogues. Eur. J. Med. Chem. 2017, 138, 371–383.
- Parrino, B.; Carbone, A.; Di Vita, G.; Ciancimino, C.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Livrea, M.; et al. 3--1H-Pyrrolo Pyridines, Nortopsentin Analogues with Antiproliferative Activity. Mar. Drugs 2015, 13, 1901–1924.
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; De Cesare, M.; et al. Novel 1 H -Pyrrolo Pyridine Derivative Nortopsentin Analogues: Synthesis and Antitumor Activity in Peritoneal Mesothelioma Experimental Models. J. Med. Chem. 2013, 56, 7060–7072.
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spano, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and Antiproliferative Activity of Substituted 3-1H-Pyrrolo Pyridines, Marine Alkaloid Nortopsentin Analogues. Curr. Med. Chem. 2014, 21, 1654–1666.
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs 2019, 17, 35.
- Spanò, V.; Attanzio, A.; Cascioferro, S.; Carbone, A.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs. Mar. Drugs 2016, 14, 226.
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Cho, H.Y.; Jung, J.H. Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from a Marine Sponge Spongosorites Sp. J. Nat. Prod. 2007, 70, 2–8.
- Kim, G.; Cheong, O.; Bae, S.; Shin, J.; Lee, S. 6″-Debromohamacanthin A, a Bis (Indole) Alkaloid, Inhibits Angiogenesis by Targeting the VEGFR2-Mediated PI3K/AKT/mTOR Signaling Pathways. Mar. Drugs 2013, 11, 1087–1103.
- Jiang, B.; Gu, X.-H. Syntheses and Cytotoxicity Evaluation of Bis(Indolyl)Thiazole, Bis(Indolyl)Pyrazinone and Bis(Indolyl)Pyrazine: Analogues of Cytotoxic Marine Bis(Indole) Alkaloid. Bioorg. Med. Chem. 2000, 8, 363–371.
- Kohmoto, S.; Kashman, Y.; Mcconnell, O.J.; Rinehart, K.L.J.; Wright, A.; Koehn, F. ChemInform Abstract: Dragmacidin, a New Cytotoxic Bis(Indole) Alkaloid from a Deep Water Marine Sponge, Dragmacidon sp. ChemInform 1988, 19.
- Morris, S.A.; Andersen, R.J. ChemInform Abstract: Brominated Bis(Indole) Alkaloids from the Marine Sponge Hexadella sp. ChemInform 1990, 21.
- Cruz, P.G.; Martínez Leal, J.F.; Daranas, A.H.; Pérez, M.; Cuevas, C. On the Mechanism of Action of Dragmacidins I and J, Two New Representatives of a New Class of Protein Phosphatase 1 and 2A Inhibitors. ACS Omega 2018, 3, 3760–3767.
- Wright, A.; Killday, K.; Chakrabarti, D.; Guzmán, E.; Harmody, D.; McCarthy, P.; Pitts, T.; Pomponi, S.; Reed, J.; Roberts, B.; et al. Dragmacidin G, a Bioactive Bis-Indole Alkaloid from a Deep-Water Sponge of the Genus Spongosorites. Mar. Drugs 2017, 15, 16.
- Jiang, B. Synthesis and Cytotoxicity Evaluation of Novel Indolylpyrimidines and Indolylpyrazines as Potential Antitumor Agents. Bioorg. Med. Chem. 2001, 9, 1149–1154.
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer Activity of Fascaplysin against Lung Cancer Cell and Small Cell Lung Cancer Circulating Tumor Cell Lines. Mar. Drugs 2018, 16, 383.
- Bryukhovetskiy, I.; Lyakhova, I.; Mischenko, P.; Milkina, E.; Zaitsev, S.; Khotimchenko, Y.; Bryukhovetskiy, A.; Polevshchikov, A.; Kudryavtsev, I.; Khotimchenko, M.; et al. Alkaloids of Fascaplysin Are Effective Conventional Chemotherapeutic Drugs, Inhibiting the Proliferation of C6 Glioma Cells and Causing Their Death in Vitro. Oncol. Lett. 2017, 13, 738–746.
- Chen, S.; Guan, X.; Wang, L.-L.; Li, B.; Sang, X.-B.; Liu, Y.; Zhao, Y. Fascaplysin Inhibit Ovarian Cancer Cell Proliferation and Metastasis through Inhibiting CDK4. Gene 2017, 635, 3–8.
- Mahgoub, T.; Eustace, A.J.; Collins, D.M.; Walsh, N.; O’Donovan, N.; Crown, J. Kinase Inhibitor Screening Identifies CDK4 as a Potential Therapeutic Target for Melanoma. Int. J. Oncol. 2015, 47, 900–908.
- Oh, T.-I.; Lee, J.; Kim, S.; Nam, T.-J.; Kim, Y.-S.; Kim, B.; Yim, W.; Lim, J.-H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules 2017, 23, 42.
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk Between Apoptosis and Autophagy Through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997.
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy Represses Fascaplysin-Induced Apoptosis and Angiogenesis Inhibition via ROS and P8 in Vascular Endothelia Cells. Biomed. Pharmacother. 2019, 114, 108866.
- Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.; Kim, Y.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074.
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the Marine Alkaloids 6-Oxofascaplysin, Fascaplysin and Their Derivatives. Tetrahedron Lett. 2018, 59, 708–711.
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor Activity of Fascaplysin Derivatives on Glioblastoma Model In Vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672.
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A Marine Sponge Alkaloid Derivative 4-Chloro Fascaplysin Inhibits Tumor Growth and VEGF Mediated Angiogenesis by Disrupting PI3K/Akt/mTOR Signaling Cascade. Chem. Biol. Interact. 2017, 275, 47–60.
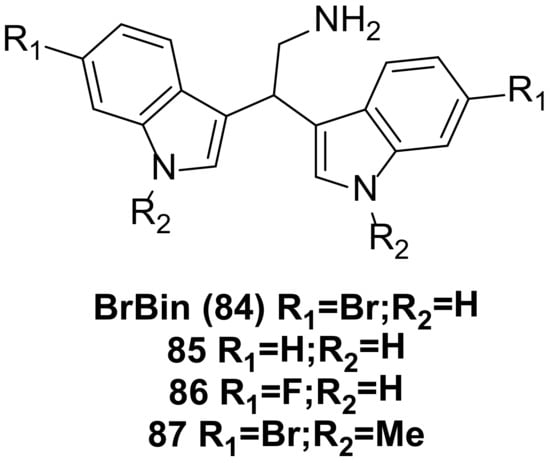
- Mantenuto, S.; Lucarini, S.; De Santi, M.; Piersanti, G.; Brandi, G.; Favi, G.; Mantellini, F. One-Pot Synthesis of Biheterocycles Based on Indole and Azole Scaffolds Using Tryptamines and 1,2-Diaza-1,3-Dienes as Building Blocks: One-Pot Synthesis of Biheterocycles Based on Indole and Azole Scaffolds Using Tryptamines and 1,2-Diaza-1,3-Dienes as Building Blocks. Eur. J. Org. Chem. 2016, 2016, 3193–3199.
- Mari, M.; Tassoni, A.; Lucarini, S.; Fanelli, M.; Piersanti, G.; Spadoni, G. Brønsted Acid Catalyzed Bisindolization of α-Amido Acetals: Synthesis and Anticancer Activity of Bis(Indolyl)Ethanamino Derivatives: Brønsted Acid Catalyzed Bisindolization of α-Amido Acetals. Eur. J. Org. Chem. 2014, 2014, 3822–3830.
- Salucci, S.; Burattini, S.; Buontempo, F.; Orsini, E.; Furiassi, L.; Mari, M.; Lucarini, S.; Martelli, A.M.; Falcieri, E. Marine Bisindole Alkaloid: A Potential Apoptotic Inducer in Human Cancer Cells. Eur. J. Histochem. 2018, 62, 2881.
- Burattini, S.; Battistelli, M.; Verboni, M.; Falcieri, E.; Faenza, I.; Lucarini, S.; Salucci, S. Morpho-functional Analyses Reveal That Changes in the Chemical Structure of a Marine Bisindole Alkaloid Alter the Cytotoxic Effect of Its Derivatives. Microsc. Res. Tech. 2022, 85, 2381–2389.
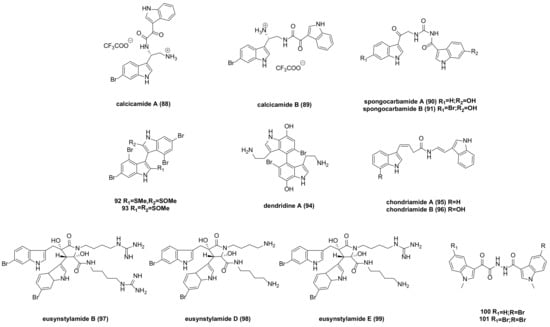
- Jennings, L.K.; Khan, N.M.D.; Kaur, N.; Rodrigues, D.; Morrow, C.; Boyd, A.; Thomas, O.P. Brominated Bisindole Alkaloids from the Celtic Sea Sponge Spongosorites Calcicola. Molecules 2019, 24, 3890.
- Park, J.S.; Cho, E.; Hwang, J.-Y.; Park, S.C.; Chung, B.; Kwon, O.-S.; Sim, C.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Bioactive Bis(Indole) Alkaloids from a Spongosorites sp. Sponge. Mar. Drugs 2020, 19, 3.
- El-Gamal, A.A.; Wang, W.-L.; Duh, C.-Y. Sulfur-Containing Polybromoindoles from the Formosan Red Alga Laurencia b Rongniartii. J. Nat. Prod. 2005, 68, 815–817.
- Tsuda, M.; Takahashi, Y.; Fromont, J.; Mikami, Y.; Kobayashi, J. Dendridine A, a Bis-Indole Alkaloid from a Marine Sponge Dictyodendrilla Species. J. Nat. Prod. 2005, 68, 1277–1278.
- Palermo, J.A.; Flower, P.B.; Seldes, A.M. Chondriamides A and B, New Indolic Metabolites from the Red Alga Chondria Sp. Tetrahedron Lett. 1992, 33, 3097–3100.
- Liberio, M.; Sadowski, M.; Nelson, C.; Davis, R. Identification of Eusynstyelamide B as a Potent Cell Cycle Inhibitor Following the Generation and Screening of an Ascidian-Derived Extract Library Using a Real Time Cell Analyzer. Mar. Drugs 2014, 12, 5222–5239.
- Tadesse, M.; Tabudravu, J.N.; Jaspars, M.; Strøm, M.B.; Hansen, E.; Andersen, J.H.; Kristiansen, P.E.; Haug, T. The Antibacterial Ent-Eusynstyelamide B and Eusynstyelamides D, E, and F from the Arctic Bryozoan Tegella Cf. Spitzbergensis. J. Nat. Prod. 2011, 74, 837–841.
- Sreenivasulu, R.; Durgesh, R.; Jadav, S.S.; Sujitha, P.; Ganesh Kumar, C.; Raju, R.R. Synthesis, Anticancer Evaluation and Molecular Docking Studies of Bis(Indolyl) Triazinones, Nortopsentin Analogs. Chem. Pap. 2018, 72, 1369–1378.
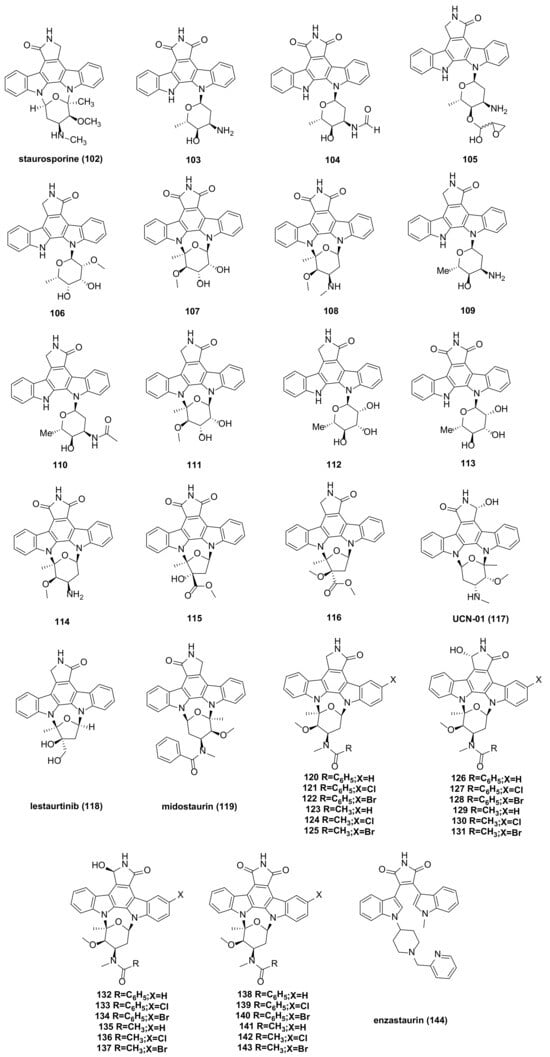
- Gayler, K.M.; Kong, K.; Reisenauer, K.; Taube, J.H.; Wood, J.L. Staurosporine Analogs Via C–H Borylation. ACS Med. Chem. Lett. 2020, 11, 2441–2445.
- Alsamman, K.; El-Masry, O. Staurosporine Alleviates Cisplatin Chemoresistance in Human Cancer Cell Models by Suppressing the Induction of SQSTM1/P62. Oncol. Rep. 2018, 40, 2157–2162.
- Malsy, M.; Bitzinger, D.; Graf, B.; Bundscherer, A. Staurosporine Induces Apoptosis in Pancreatic Carcinoma Cells PaTu 8988t and Panc-1 via the Intrinsic Signaling Pathway. Eur. J. Med. Res. 2019, 24, 5.
- Ding, Y.; Wang, B.; Chen, X.; Zhou, Y.; Ge, J. Staurosporine Suppresses Survival of HepG2 Cancer Cells through Omi/HtrA2-Mediated Inhibition of PI3K/Akt Signaling Pathway. Tumor Biol. 2017, 39, 101042831769431.
- Chen, Z.; Zhou, K.; Xue, J.; Small, A.; Xiao, G.; Nguyen, L.X.T.; Zhang, Z.; Prince, E.; Weng, H.; Huang, H.; et al. Phosphorylation Stabilized TET1 Acts as an Oncoprotein and Therapeutic Target in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2023, 15, eabq8513.
- Zhou, B.; Hu, Z.-J.; Zhang, H.-J.; Li, J.-Q.; Ding, W.-J.; Ma, Z.-J. Bioactive Staurosporine Derivatives from the Streptomyces sp. NB-A13. Bioorganic Chem. 2019, 82, 33–40.
- Kurata, N.; Kuwabara, T.; Tanii, H.; Fuse, E.; Akiyama, T.; Akinaga, S.; Kobayashi, H.; Yamaguchi, K.; Kobayashi, S. Pharmacokinetics and Pharmacodynamics of a Novel Protein Kinase Inhibitor, UCN-01. Cancer Chemother. Pharmacol. 1999, 44, 12–18.
- Zhao, B.; Bower, M.J.; McDevitt, P.J.; Zhao, H.; Davis, S.T.; Johanson, K.O.; Green, S.M.; Concha, N.O.; Zhou, B. Structural Basis for Chk1 Inhibition by UCN-01 and Its Analogs. Acta Crystallogr. A 2002, 58, c224.
- Sato, S.; Fujita, N.; Tsuruo, T. Interference with PDK1-Akt Survival Signaling Pathway by UCN-01 (7-Hydroxystaurosporine). Oncogene 2002, 21, 1727–1738.
- Lien, W.; Chen, T.; Sheu, S.; Lin, T.; Kang, F.; Yu, C.; Kuan, T.; Huang, B.; Wang, C. 7-hydroxy-staurosporine, UCN-01, Induces DNA Damage Response, and Autophagy in Human Osteosarcoma U2-OS Cells. J. Cell. Biochem. 2018, 119, 4729–4741.
- Candido, M.F.; Medeiros, M.; Veronez, L.C.; Bastos, D.; Oliveira, K.L.; Pezuk, J.A.; Valera, E.T.; Brassesco, M.S. Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario. Pharmaceutics 2023, 15, 664.
- Arena, A.; Romeo, M.A.; Benedetti, R.; Gilardini Montani, M.S.; Cirone, M. The Impairment of DDR Reduces XBP1s, Further Increasing DNA Damage, and Triggers Autophagy via PERK/eIF2alpha in MM and IRE1alpha/JNK1/2 in PEL Cells. Biochem. Biophys. Res. Commun. 2022, 613, 19–25.
- Levis, M.; Allebach, J.; Tse, K.-F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-Targeted Tyrosine Kinase Inhibitor Is Cytotoxic to Leukemia Cells in Vitro and in Vivo. Blood 2002, 99, 3885–3891.
- Armstrong, S.A.; Kung, A.L.; Mabon, M.E.; Silverman, L.B.; Stam, R.W.; Den Boer, M.L.; Pieters, R.; Kersey, J.H.; Sallan, S.E.; Fletcher, J.A.; et al. Inhibition of FLT3 in MLL. Cancer Cell 2003, 3, 173–183.
- Azam, H.; Pierro, L.; Reina, M.; Gallagher, W.M.; Prencipe, M. Emerging Role for the Serum Response Factor (SRF) as a Potential Therapeutic Target in Cancer. Expert Opin. Ther. Targets 2022, 26, 155–169.
- Fathi, A.T.; Levis, M. Lestaurtinib: A Multi-Targeted FLT3 Inhibitor. Expert Rev. Hematol. 2009, 2, 17–26.
- Stansfield, L.C.; Pollyea, D.A. Midostaurin: A New Oral Agent Targeting FMS -Like Tyrosine Kinase 3-Mutant Acute Myeloid Leukemia. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 1586–1599.
- Kim, E.S. Midostaurin: First Global Approval. Drugs 2017, 77, 1251–1259.
- Levis, M. Midostaurin Approved for FLT3-Mutated AML. Blood 2017, 129, 3403–3406.
- Wang, L.; Zhuang, Y.; Sun, K.; Zhu, W. Synthesis and Cytotoxicity of Halogenated Derivatives of PKC-412. Chin. J. Org. Chem. 2014, 34, 1603.
- Wang, L.; Mei, X.; Wang, C.; Zhu, W. Biomimetic Semi-Synthesis of Fradcarbazole A and Its Analogues. Tetrahedron 2015, 71, 7990–7997.
- Li, M.; Xu, Y.; Zuo, M.; Liu, W.; Wang, L.; Zhu, W. Semisynthetic Derivatives of Fradcarbazole A and Their Cytotoxicity against Acute Myeloid Leukemia Cell Lines. J. Nat. Prod. 2019, 82, 2279–2290.
- Bourhill, T.; Narendran, A.; Johnston, R.N. Enzastaurin: A Lesson in Drug Development. Crit. Rev. Oncol. Hematol. 2017, 112, 72–79.


