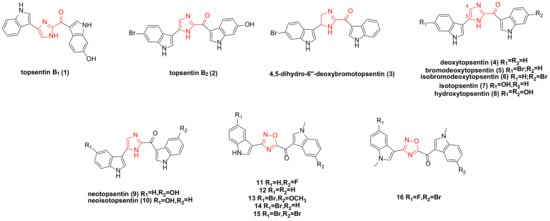
1. Topsentin Family Bisindole Alkaloids and Their Derivatives
Topsentins family bisindole alkaloids are compounds that utilize carbonyl imidazole as a spacer to link two indole moieties, predominantly isolated from deep-sea sponges.
A research team isolated topsentin B
1 (
1) (
Figure 1) from a sponge, which exhibited significant cytotoxicity to the P388 cancer cell line with an IC
50 of 4.1 ± 1.4 µM. It also demonstrated a moderate inhibitory effect on human HL-60 cancer cells, with an IC
50 of 15.7 ± 4.3 µM, and exhibited anti-proliferative effects on human tumor cells HCT-8, A-549, and T47D. In vivo tumor models established in mice confirmed that topsentin B
1 had an inhibitory effect on P388 and B16. Further research on P388 cancer cells revealed that the compound could inhibit DNA (deoxyribonucleic acid) (91%) and RNA (ribonucleic acid) synthesis (57%), while not affecting protein synthesis
[1][2][12,13].
Figure 1. Topsentin family bisindole alkaloids and their derivatives.
Topsentin B
1 (
1) and topsentin B
2 (
2) (
Figure 1) exhibited moderate anti-proliferative activity against NSCLC-N6 (non-small cell lung cancer), with IC
50 values of 12 and 6.3 µg/mL, respectively. They also displayed cytotoxicity to HeLa cells, with IC
50 values of 4.4 and 1.7 µM, respectively
[3][4][5][14,15,16]. (Topsentin B
2, also known as bromotopsentin).
The four bisindole alkaloids topsentin B
2 (
2), 4,5-dihydro-6″-deoxybromotopsentin (
3), deoxytopsentin (
4), and bromodeoxytopsentin (
5) (
Figure 1), extracted from the sponge Spongosorites sp., all showed moderate activity (IC
50: 1.1–7.4 µg/mL) against AGS (adenocarcinoma) and L1210 (lymphocytic leukemia). Compounds
4 and
3 demonstrated moderate anti-proliferative activity on BC (breast cancer) cancer cells, with IC
50 values of 10.7 and 17.5 µg/mL, respectively. Compound
4 also exhibited moderate activity against HepG2, with an IC
50 of 3.3 µg/mL
[6][17]. It was previously reported that topsentin B
2 had cytotoxicity to P388 cells, with an IC
50 of 7.0 µg/mL
[7][18].
Bao’s research team isolated bromodeoxytopsentin (
5) and isobromodeoxytopsentin (
6) (
Figure 1) from marine sponge sp., which were cytotoxic to K562 cells with IC
50 values of 0.6 and 2.1 µg/mL, respectively
[8][19]. Compound
6 is cytotoxic to five human cancer cell lines: A549, SK-OV-3, SK-MEL-2, XF498, and HCT15
[9][20].
In 1988, while extracting natural products from topsentins, several analogues were synthesized. Under identical conditions, the cytotoxicity of natural products
1–
4 and analogues
7–
10 (
Figure 1) was evaluated. The IC
50 values for natural products
1–
4 against P388 cancer cells were 2.0, 12.0, 4.0, and 7.0 µg/mL, respectively, while those for analogues
7–
10 were 4.0, 0.3, 2.5, and 1.8 µg/mL, respectively. The results indicated that the introduction of a hydroxyl group enhanced cytotoxicity, whereas the introduction of a bromine substituent reduced it
[1][12].
Azaindole and its derivatives have demonstrated significant biological activities in previous studies, including antitumor effects
[10][11][12][13][14][21,22,23,24,25]. A team reported synthesizing a series of new 1,2,4-oxadiazole topsentin analogues and screened five compounds,
11–
15 (
Figure 1), which showed strong activity on pancreatic ductal adenocarcinoma cell lines (including Panc-1, Capan-1, and SUIT-2), with EC
50 values ranging from 0.4 to 6.8 µM. None of these derivatives exhibited cytotoxic effects against non-tumor pancreatic cells. Further studies revealed that the anti-proliferation mechanism of compounds
11–
15 involved promoting apoptosis, which was related to the externalization of phosphatidylserine in the cell membrane, suggesting that their target was GSK3β (glycogen synthase kinase 3 beta)
[15][26].
The existence of a carbonyl spacer group in the topsentin family allows small-molecule compounds to have greater flexibility, better adapt to the ATP (adenosine triphosphate) binding site of CDK1 (cyclin-dependent kinase 1), and act as a hydrogen bond receptor. This enables interaction with amino acid residues at the active site of CDK1. The designed and synthesized derivative
16 (
Figure 1) exhibited a significant anti-proliferative effect against PDAC (pancreatic ductal adenocarcinoma cells) (Hs766T, HPAF-II, PDAC3, and PATU-T), with IC
50 values of 5.7 ± 0.60, 6.9 ± 0.25, 9.8 ± 0.70, and 10.7 ± 0.16 µM, respectively. This compound promoted apoptosis in PDAC cells and regulated the expression of CDK1. ADME (absorption, distribution, metabolism, and excretion) prediction indicated good pharmacokinetic properties, warranting further clinical investigation
[16][27].
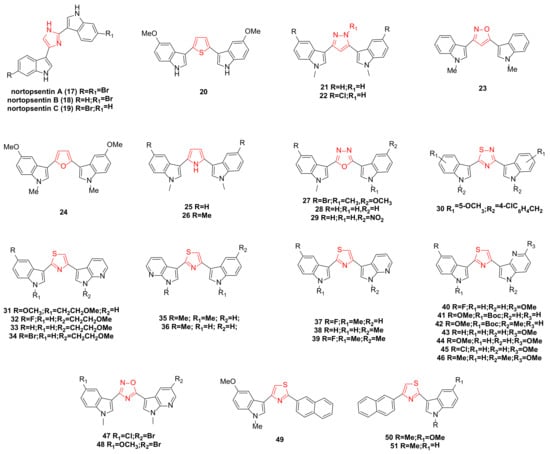
2. Nortopsentin Family Bisindole Alkaloids and Their Derivatives
Nortopsentins A–C (
17–
19) (
Figure 2) possess a 2,4-bis(3′-indolyl)imidazole structure skeleton, which exhibits significant inhibitory effects against P338 cells, with IC
50 values of 7.6, 7.8, and 1.7 µg/mL, respectively
[7][18]. In recent years, the unique structure and anticancer potential of nortopsentins bisindole alkaloids have garnered the attention of many research teams, leading to a series of modifications. The modifications primarily target the middle part (five-membered heterocycle) of the two indole structures and further extend to one or two indole units
[17][28].
Figure 2. Noropsentin family bisindole alkaloids and their derivatives.
Patrizia Diana’s team reported the synthesis of bisindole thiophene and bisindole pyrazole derivatives. Several of these compounds were selected for evaluation against 60 cancer cell lines from nine human cancer types. The results indicated that the most active bisindole thiophene compound was
20 (
Figure 2), particularly effective against leukemic cell lines (CCRF-CEM, MOLT-4, HL-60, RPMI-8226, K-562) with GI
50 values ranging from 0.34 to 3.54 µM. It demonstrated good selectivity for the HT29 and HCC-2998 cell lines of the colon cancer subgroup, with GI
50 values of 2.79 and 2.83 µM, respectively. It also showed selectivity for NCI-H522 of the non-small cell lung cancer subgroup, LOXIMVI of the melanoma subgroup, and UO31 of the renal cancer subgroup
[18][29]. Bisindole pyrazole
21 (
Figure 2) exhibited antitumor activity on most human cell lines, while compound
22 (
Figure 2), containing more chlorine groups than compound
21, was effective on all tested cells
[19][30]. The team also synthesized a series of 2,5-bis(3′-indolyl)furan and 3,5-bis(3′-indolyl)isoxazole antitumor drugs. The in vitro antitumor activities of these compounds against 10 human tumor cells were tested, revealing that all compounds exhibited antiproliferative activity at the highest test concentration of 100 µg/mL. Further tests on compounds
23 and
24 (
Figure 2), which showed strong activities in 29 cell lines, revealed that compound
24 exhibited a high level of tumor selectivity, and compound
23 demonstrated selective activity against A549, LXFA629L, and UXF1138L
[20][31].
A series of compounds were synthesized by replacing the imidazole ring with a pyrrole ring. All compounds exhibited anti-proliferative activity at the highest test concentration of 100 µg/mL. Among them, compounds
25 and
26 (
Figure 2) demonstrated the most significant anti-proliferative activity against a group of human tumor cell lines (42 human tumor cells) cultured in vitro, with an average IC
50 value of 0.37 µg/mL. Moreover, in vitro clonogenic assays revealed significant tumor selectivity for both compounds, particularly for compound
26 [21][32].
New 2,5-bisindolyl-1,3,4-oxadiazoles were designed and synthesized. Compounds
27 and
29 (
Figure 2) exhibited strong anticancer activity against the MCF-7 cell line, with IC
50 values of 1.8 ± 0.9 and 2.6 ± 0.89 µM, respectively. Compounds
27,
28, and
29 (
Figure 2) displayed potent cytotoxicity against HeLa cells, with IC
50 values of 9.23 ± 0.58, 9.4 ± 0.37, and 6.34 ± 0.56 µM, respectively. Notably, compound
29 demonstrated significant cytotoxicity against the lung cancer cell line A549, with an IC
50 value of 3.3 ± 0.85 µM. Importantly, these compounds showed no cytotoxicity against normal human embryonic kidney cells, HEK-293
[22][33].
A series of bisindole thiadiazole compounds was synthesized and tested for in vitro cytotoxicity against six human cancer cell lines: prostate (PC3, DU145, and LNCAP), breast (MCF-7 and MDA-MB-231), and pancreas (PACA2). The results indicated that substituents at the N-1 and C-6 positions of the indole ring were crucial for inducing cytotoxicity and selectivity towards specific cancer cell lines. Among them, compound
30 (
Figure 2), with a 4-chlorobenzyl group and a 5-methoxy substituent, showed significant inhibitory effects on all cancer cell lines tested (IC
50 range: 14.6–369.8 µM), particularly on LNCAP cancer cell lines (IC
50 = 14.6 µM)
[23][34].
With further research, a series of new thiazole nortopsentin analogues were synthesized. To increase water solubility, the nitrogen atom of the indole, or 7-azaindole, was modified with a 2-methoxyethyl chain. Among these, four derivatives (
31–
34) (
Figure 2) demonstrated potent anti-proliferative activity against nearly all 60 human tumor cell lines (GI
50 range: 0.03–98.0 µM). Further studies revealed that the anti-proliferation mechanism of these compounds against MCF-7 cells involved promoting apoptosis, associated with the externalization of phosphatidylserine in the cell membrane and DNA fragmentation. Compound
31, exhibiting the strongest activity and selectivity, constrained living cells to the G2/M phase and significantly inhibited the activity of CDK1 in vitro
[24][35].
Compounds
35 and
36 (
Figure 2) displayed anti-proliferative activity against about 60 human tumor cell lines, with GI
50 values of 0.03–13.0 µM and 0.04–14.2 µM, respectively, and did not significantly affect the survival rate of normal human liver cells, indicating high selectivity towards tumor cells. After treating HepG2 cells with these compounds, cell cycle distribution studies showed concentration-dependent accumulation in the sub-G0/G1 phase, restricting living cells to the G2/M phase. The anti-proliferation mechanism involved promoting apoptosis, linked to the externalization of phosphatidylserine in the cell membrane and mitochondrial dysfunction
[25][36].
Compounds
37,
38, and
39 (
Figure 2) significantly inhibited the activity of CDK1, with IC
50 values of 0.89 ± 0.07, 0.75 ± 0.03, and 0.86 ± 0.04 µM, respectively, and did not substantially interfere with the proliferation of human normal fibroblasts. In a nude mouse tumor model, these three compounds were also found to have significant inhibitory effects on tumor cell proliferation and growth. Further investigation into their mechanism of action revealed that they could block the cell cycle in the G2/M phase by inhibiting CDK1 activity, increasing the rate of apoptosis, and reducing the phosphorylated form of the anti-apoptotic protein survivin. Additionally, compound
38 synergized with paclitaxel, significantly inhibiting the survival of DMPM (diffuse malignant peritoneal mesothelioma) cells
[26][37].
A series of seven newly synthesized compounds,
40–
46 (
Figure 2), inhibited the growth of four cancer cell lines: MDA-MB-231, MIAPACA-2, PC3, and STO. Among these, compound
45 exhibited the highest activity against malignant peritoneal mesothelioma (STO) cell lines. Further investigation into the mechanism of compound
45 revealed that its treatment of STO cells significantly reduced CDK1 activity in a time-dependent manner and could induce cell cycle arrest in the G2/M phase, accompanied by marked caspase-dependent apoptosis. The phosphorylated form of the anti-apoptotic protein survivin also showed a time-dependent decrease. Additionally, compound
45 synergized with paclitaxel due to the enhanced induction of caspase-3-mediated apoptosis
[27][38].
A new nortopsentin analog was synthesized by partially replacing the central imidazole ring with 1,2,4-oxadiazole and an indole moiety with 7-azaindole. Among the synthesized derivatives, compounds
47 and
48 (
Figure 2) exhibited significant cytotoxicity against MCF-7, HCT-116, and HeLa cells (IC
50: 0.65–13.96 µM). Further studies on the anti-proliferation mechanism in MCF-7 cells showed that they could promote apoptosis, which was associated with the externalization of phosphatidylserine in the cell membrane, chromatin condensation, and membrane blebbing. They also induced cell accumulation in the G0-G1 phase
[28][39].
A team synthesized a new thiazole nortopsentin analog in which the naphthyl group partially replaced the original indole part and the other indole was replaced or retained by 7-azaindole. Three derivatives,
49–
51 (
Figure 2), demonstrated effective anti-proliferative activity against MCF-7 cells, with IC
50 values of 2.13 ± 0.12, 3.26 ± 0.19, and 5.14 ± 0.34 µM, respectively. Further cytotoxicity studies on MCF-7 cells showed that all three compounds promoted apoptosis, inducing cells to transition towards early apoptosis without necrosis. They also caused cell cycle disruption, significantly reducing the percentage of G0/G1 and S phase cells, increasing the percentage of G2/M phase cells, and leading to the appearance of a sub-G1 cell population
[29][40].
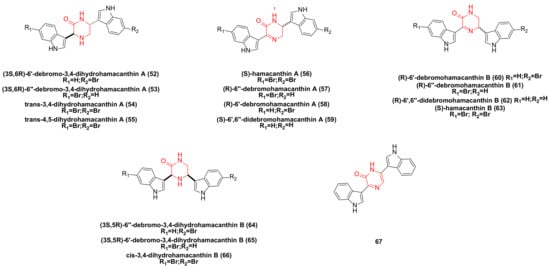
3. Hamacanthin Family Bisindole Alkaloids and Their Derivatives
Bao’s research team isolated the hamacanthin family of bisindole alkaloids from marine sponges sp. Compounds such as (3S,6R)-6′-debromo-3,4-dihydrohamacanthin A (
52), (3S,6R)-6″-debromo-3,4-dihydrohamacanthin A (
53), trans-3,4-dihydrohamacnathin A (
54), (S)-hamacanthin A (
56), (R)-6″-debromohamacanthin A (
57), (S)-6′,6″-didebromohamacanthin A (
59), (R)-6′-debromohamacanthin B (
60), (R)-6″-debromohamacanthin B (
61), (R)-6′,6″-didebromohamacanthin B (
62), (S)-hamacanthin B (
63), (3S,5R)-6″-debromo-3,4-dihydrohamacanthin B (
64), (3S,5R)-6′-debromo-3,4- dihydrohamacanthin B (
65), cis-3,4-dihydrohamacanthin B (
66) (
Figure 3) is cytotoxic to five human cancer cell lines: A549, SK-OV-3, SK-MEL-2, XF498, and HCT15. (R)-6′-debromohamacanthin A (
58) (
Figure 3) exhibited a weak inhibitory effect against HCT15
[8][9][30][19,20,41]. A study has shown that compound
58 can inhibit VEGF (vascular endothelial growth factor)-induced expression of MAPKs (p38, ERK, and SAPK/JNK) and the PI3K/AKT/mTOR signaling pathway
[31][42]. (Note: the naming of compounds
57 and
58 here is subject to reference
[9][20]).
Figure 3. Hamacanthin family bisindole alkaloids and their derivatives.
The bisindole alkaloids trans-4,5-dihydrohamacanthin A (
55), hamacanthin A (
56),
57, 6″-debromohamacanthin B (
61), and hamacanthin B (
63) were extracted from the sponge Spongosorites sp. These compounds demonstrated moderate activity against AGS and L1210 (IC
50: 1.1–9.0 µg/mL)
[6][17].
The pyrazinone bisindole compound
67 (
Figure 3) synthesized by Jiang’s team demonstrated a strong inhibitory effect on a variety of tumor cell lines, including leukemia, non-small cell lung cancer, colon cancer, CNS (central nervous system) cancer, ovarian cancer, kidney cancer, and breast cancer cell lines, with GI
50 values ranging from 6.6–74.8 µM
[32][43].
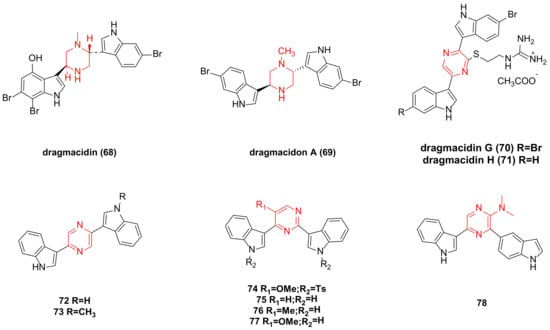
4. Dragmacidin Family Bisindole Alkaloids and Their Derivatives
The IC
50 value of dragmacidin (
68) (
Figure 4) extracted from deep-sea sponge against P388 cells is 15 µg/mL, and against A-549, HCT-8, and MDAMB cells is 1–10 µg/mL
[33][44].
Figure 4. Dragmacidin family bisindole alkaloids and their derivatives.
Dragmacidon A (
69) (
Figure 4) was isolated from the Pacific sponge
Hexadella sp. In vitro cytotoxicity tests revealed that
69 exhibited cytotoxicity in the L1210 assay (ED
50 was 10 mg/mL)
[34][45]. Dragmacidon A has significant anti-cancer activity against A549, HT29, and MDA-MB-231, with IC
50 values of 3.1, 3.3, and 3.8 µM, respectively. Research has shown that it can inhibit the mitosis of tumor cells by inhibiting PP1 and/or PP2A phosphatase. This may represent a new paradigm to inhibit Ser-Thr PPs type 1 and possibly other analogous phosphatases such as PP2A
[35][46].
Dragmacidin G (
70) (
Figure 4), composed of a pyrazine ring and two indole rings, was isolated from sponges of the genus Spongosorites. In tests using PANC-1, MIA PaCa-2, BxPC-3, and ASPC-1 cancer cell lines, the IC
50 values after 72 h of treatment were 18 ± 0.4 µM, 26 ± 1.4 µM, 14 ± 1.4 µM, and 27 ± 0.8 µM, respectively
[36][47]. Other studies also indicated that dragmacidins G (
70) and H (
71) (
Figure 4) showed moderate cytotoxicity against HeLa cells, with IC
50 values of 4.2 and 4.6 µM, respectively
[5][16].
Synthetic pyrazine bisindole compounds
72 and
73 (
Figure 4) were tested, and although compound
72 showed weak cytotoxicity, its N-methylated derivative
73 exhibited significant inhibitory activity against leukemia, non-small cell lung cancer, colon cancer, CNS cancer, ovarian cancer, kidney cancer, and breast cancer cell lines, with GI
50 values ranging from 0.058 to 7.19 µM
[32][43].
New pyrimidine bisindole and pyrazine bisindole compounds were synthesized as potential anticancer drugs. When screened across 60 in vitro human tumor cell lines (encompassing leukemia, non-small cell lung cancer, colon cancer, CNS cancer, ovarian cancer, kidney cancer, and breast cancer cell lines), compounds
74,
76,
77, and
78 (
Figure 4) exhibited notable cytotoxicity in the low micromolar range, with GI
50 values less than 11.7 µM. The cytotoxicity of compound
74 was the most pronounced, with GI
50 values ranging from 0.16 to 1.42 µM. Compound
75 showed selective inhibitory effects against the IGROV1 cell line
[37][48].
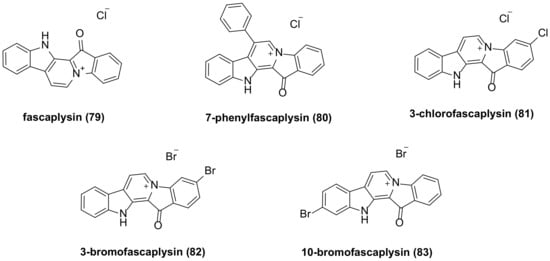
5. Fascaplysin Family Bisindole Alkaloids and Their Derivatives
The researchers isolated fascaplysin (
79) (
Figure 5) from the Fijian sponge Fascaplysinopsis
Bergquist sp.
[14][25]. Fascaplysin exhibits a broad range of anticancer activities against various cancer cell types, including NSCLC and SCLC (small cell lung cancer) cancer cells
[38][49], glioma cells
[39][50], A2780 and OVCAR3 human ovarian cancer cell lines
[40][51], melanoma cells
[41][52], and others. Fascaplysin exhibits anti-cancer effects in various types of cancer cells by inhibiting CDK4 (cyclin-dependent kinase 4)-mediated cell cycle progression. It helps to improve the anticancer effect of PI3K-AKT-targeted drugs, and when used in combination with AMPK (Adenosine 5′-monophosphate-activated protein kinase) inhibitors, it can also effectively inhibit tumor growth. Fascaplysin inhibits the expression of several folate and purine metabolism-related genes, such as MTR (5-methyltetrahydrofolate-homocysteine methyltransferase) and DHFR (dihydrofolate reductase), and makes cells sensitive to MTX (methotrexate)-induced apoptosis
[42][53]. A research team has found that fascaplysin can induce HL-60 cell death through a synergistic effect between apoptosis and autophagy. The role of autophagy is activated by inhibiting the PI3K/Akt/mTOR signaling pathway, leading to increased expression of LC3-II, ATG7, and Beclin-1
[43][54]. Fascaplysin can trigger autophagy in endothelial cells by activating the ROS pathway
[44][55]. Fascaplysin can inhibit the expression of survivin. In malignant melanoma, colorectal cancer, and lung cancer cells, fascaplysin can significantly increase TRAIL (TNF-related apoptosis-inducing ligand)-induced cancer cell death. These results suggest that fascaplysin may be a potential drug to overcome resistance to TRAIL-based cancer therapies
[45][56].
Figure 5. Fascaplysin family bisindole alkaloids and their derivatives.
The in vitro antiproliferative activity of the synthesized fascaplysin derivative
80 (
Figure 5) demonstrated IC
50 values of 290 nM and 320 nM against HeLa (cervical cancer) and THP-1 (acute monocytic leukemia) cell lines, respectively, while the IC
50 values of fascaplysin were 550 nM and 890 nM, respectively. This indicates that the inhibitory activity of fascaplysin derivatives with phenyl substituents at the C-7 position on selected cancer cell lines is 2–3 times higher than that of fascaplysin itself
[46][57].
Lyakhova’s team synthesized four fascaplysin derivatives,
80–
83 (
Figure 5). Cytotoxicity test results indicated that all compounds exhibited higher cytotoxicity than unsubstituted fascaplysin. Among these, 7-phenyl fascaplysin (
80) and 3-bromo fascaplysin (
82) were particularly effective against glioma C6 cells, with apoptosis being the primary mechanism of cell death. The cytotoxicity of these compounds was time- and concentration-dependent. Fascaplysin derivatives altered all stages of the glioma cell life cycle, and both significantly reduced the number of viable tumor cells in the G0 phase
[47][58]. Compound
81 inhibits VEGF (vascular endothelial growth factor)-mediated angiogenesis, induces autophagy and apoptosis, and interferes with the PI3K/Akt/mTOR signaling pathway in MDAMB-231 cells
[48][59].
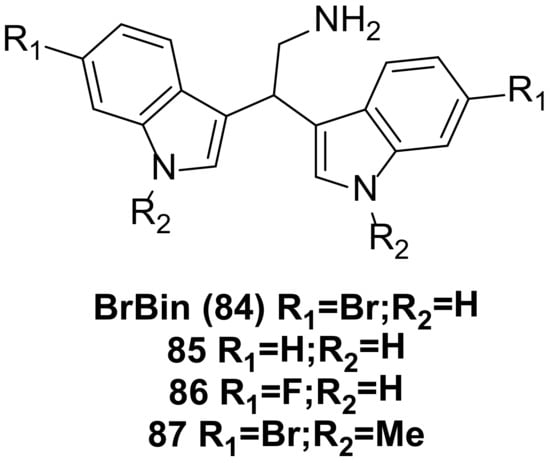
6. Bisindole Acetylamine Alkaloids and Their Derivatives
2,2-bis (6-bromo-3-indolyl) ethylamine (
84) (
Figure 6), also known as BrBin, was isolated from the new caledonian sponge Orina and showed high cytotoxicity against U937, MCF-7, and Caco-2 cancer cell lines
[49][50][60,61]. BrBin is a potent apoptotic agent. Its mechanism was first evaluated in the U937 tumor cell model. Studies revealed that BrBin modulates the ratio between anti-apoptotic and pro-apoptotic factors and induces mitochondria to participate in the activation of apoptosis mechanisms through the down-regulation of Bcl-2/Bcl-xL and the up-regulation of Bax, thus promoting apoptotic cell death in U937. The compound’s effect is dose-dependent
[51][62].
Figure 6. Bisindole acetylamine alkaloids and their derivatives.
Under similar conditions, compounds
85 and
86 (
Figure 6) reduced U937 cell viability by about 10% compared to BrBin, while compound
87 (
Figure 6) induced more cell death. Consequently, compound
87 maintained cytotoxicity equivalent to that observed after BrBin treatment. The structure-activity relationship studies indicated that the presence of a bromine atom and N-methylation are essential for apoptotic activity, and compound
87 functions in driving apoptotic cell death through mitochondrial involvement
[52][63].
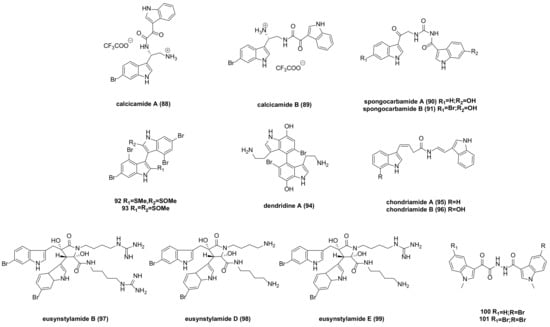
7. Acyclic Structure-Linked Bisindole Alkaloids and Their Derivatives
Calcicamide A (
88) and B (
89) (
Figure 7) were isolated from the Irish sponge S. Calcicola. The cytotoxicity of calcicamide B against HeLa cells was measured after 6 and 24 h of incubation, revealing IC
50 values of 165 ± 7 and 146 ± 13 µM, respectively, stronger than that of calcicamide A (IC
50 > 200 µM)
[53][64].
Figure 7. Acyclic structure linked bisindole alkaloids and derivatives.
Spongocarbamides A and B (
90–
91) (
Figure 7), which connect indole groups through linear urea-type bonds, exhibited weak inhibitory effects against A549 and K562 cancer cell lines, with IC
50 > 90 µM
[54][65].
Bisindoles
92 and
93 (
Figure 7) were extracted from the formosan red alga, laurencia brongniartii. Compound
92 showed an inhibitory effect against HT-29 and P388 cancer cells, while compound
93 had a cytotoxic effect on P388 cells. Specific IC
50 values are not provided in the literature
[55][66].
Dendridine A (
94) (
Figure 7) exhibited weak cytotoxicity to mouse leukemia L1210 cells, with an IC
50 of 32.5 μg/mL
[56][67].
Chondriamides A (
95) and B (
96) (
Figure 7), two cytotoxic bisindole amides, were isolated from the red alga chondria sp. Cytotoxicity tests indicated that chondriamide A had an IC
50 of 0.5 μg/mL against KB cells and 5 μg/mL against LOVO (Human Colorectal Carcinoma Cells) cells, while the activity of chondriamide B on these two cancer cell types was <1 μg/mL and 10 μg/mL, respectively
[57][68].
Eusynstyelamide B (
97) (
Figure 7), a natural product isolated from ascidians, was tested for toxicity on MDA-MB-231 cells, resulting in an IC
50 of 5.0 µM. Further cell cycle analysis revealed significant stagnation in the G2/M phase
[58][69]. Eusynstyelamides D (
98) and E (
99) (
Figure 7), obtained by acid degradation of guanidine, showed weak cytotoxicity against the melanoma cell line A-2058, with IC
50 values of 57 and 114.3 µM, respectively
[59][70].
A series of bisindolyl triazinone analogues synthesized by Reddymasu Srenivasulu’s team were tested for in vitro activity. Compounds
100 and
101 (
Figure 7) exhibited significant antiproliferative activity against human cervical cancer cell lines, with IC
50 values of 4.6 and 1.3 µM, respectively, and showed no cytotoxic effects on HEK-293 cell lines (human embryonic kidney cells)
[60][71].
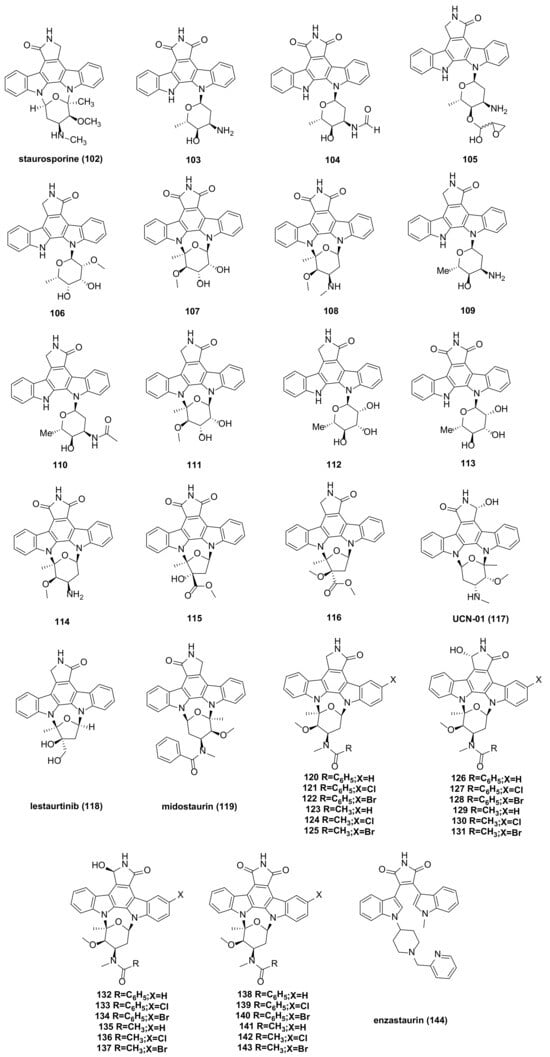
8. Indolecarbazole Bisindole Alkaloids and Their Derivatives
STU (Staurosporine) (
102) (
Figure 8), isolated from the culture medium of Streptomyces marinus, is one of the most potent natural PKC (protein kinase C) inhibitors identified to date. It has shown considerable anti-tumor potential and has been extensively studied as a lead compound
[61][72]. Staurosporine can sensitize cancer cells to cisplatin by down-regulating the p62 mechanism. In other cancer models, the combination of staurosporine and cisplatin may reduce cell proliferation and help overcome cisplatin resistance
[62][73]. It can also induce apoptosis in pancreatic cancer cells (PaTu 8988t and Panc-1) through the intrinsic signaling pathway
[63][74]. Staurosporine significantly induces autophagy by inhibiting the PI3K/Akt/mTOR signaling pathway, thereby inducing HepG2 cell death
[64][75]. The ATM (ATM Serine/Threonine Kinase) inhibitor (AZD0156) and staurosporine inhibit the phosphorylation of TET1 (tet methylcysteine dioxygenase 1), causing instability of the TET1 protein and demonstrating a synergistic killing effect on B-ALL (B cell acute lymphoblastic leukemia) cells
[65][76]. However, the poor target specificity and high toxicity of staurosporine have led to the failure of most preclinical studies.
Figure 8. Indolecarbazole bisindole alkaloids and their derivatives.
Several staurosporine derivatives were isolated from the rice solid fermentation broth of the marine-derived
Streptomyces sp. NB-A13. The inhibitory activity of compound
108 (
Figure 8) on the SW-620 cell line was stronger than that of the positive control staurosporine, with an IC
50 value of 9.99 nM, compared to staurosporine’s IC
50 of 25.10 nM. Compounds
103–
107,
109–
114, and
116 (
Figure 8) also exhibited notable cytotoxicity, with IC
50 values ranging from 0.02 to 16.60 µM. Compound
108 (
Figure 8) demonstrated strong cytotoxicity, suggesting that bridged sugar units are crucial for cytotoxicity. This was further evidenced by the increased cytotoxicity of compounds
107,
111, and
114 compared to other analogs. The PKC enzyme inhibition activity of compounds
103–
108 was tested, with IC
50 values ranging from 0.06 to 9.43 µM
[66][77].
STU analogues generally exhibit lower toxicity compared to STU itself. Several of these analogues have been used in clinical and preclinical studies, including UCN-01 (
117), lestaurtinib (
118), and midostaurin (
119) (
Figure 8). UCN-01 is a selective inhibitor of protein kinase C
[67][78] and has been shown to inhibit the protein expression of thymidylate synthase and E2F-1 (E2F transcription factor 1), as well as the DNA damage checkpoint kinase Chk1 (Check point kinase 1)
[68][79]. It mediates Akt inactivation by inhibiting the upstream Akt kinase PDK1 (3-phosphoinositide-dependent protein kinase 1)
[69][80] and can induce autophagy activation to protect cells from apoptosis, suggesting its potential as a standalone anticancer agent in treating human osteosarcoma
[70][81]. UCN-01 inhibits cell proliferation in a group of NB (neuroblastoma) cell lines and detects the induction of cell apoptosis through caspase activation, caspase-3, and PARP cleavage
[71][82]. AZD2461 (an effective PARP inhibitor)/UCN-01 combined inhibit PARP (poly (ADP ribose) polymer) and CHK1, reducing the expression of xbp1 (X-Box Binding Protein 1) in cells by downregulating c-Myc (cellular-myelocytomatosis viral oncogene) and upregulating the pro-apoptotic molecule CHOP (C/EBP-homologous protein
), leading to UPR (unfolded protein response) imbalance and cell death and trigger autophagy through PERK/eIF2alpha in MM (multiple myeloma) and IRE1alpha/JNK1/2 in PEL (primary effusion lymphoma) cells
[72][83]. Lestaurtinib is a multi-target FLT3 (Fms-like Tyrosine Kinase-3) inhibitor (
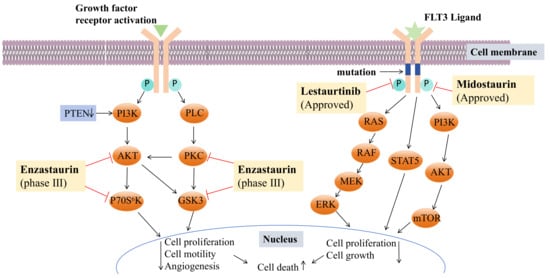
Figure 9)
[73][84] with confirmed efficacy in inhibiting phosphorylation and inducing cell death in primary leukemia samples and in vivo mouse model studies
[74][85]. In recent years, lestaurtinib has been shown to inhibit PKN1 (protein kinase N1), thereby affecting the SRF (serum response factor) activity induced by androgens in PCa (prostate cancer) cells and xenograft models. Many studies have shown that SRF plays a dominant role in the development and progression of various types of cancer
[75][86]. The development history of lestaurtinib has been described in a review by Levis
[76][87]. Midostaurin is the first oral multitargeted TKI (tyrosine kinase inhibitors) to improve overall survival in patients with FLT3-mutant AML (acute myeloid leukemia) and represents an important addition to the limited armamentarium against AML
[77][88]. The first compound performed poorly in clinical trials due to its targeting effect. Lastly, lestaurtinib and midostaurin were developed for the treatment of acute myeloid leukemia with a positive FLT3 mutation (
Figure 9)
[78][79][89,90].
Figure 9. Mechanisms of enzastaurin, midostaurin, and lestaurtinib. (FLT3: Fms-like TyrosineKinase-3, PKC: protein kinase C, PTEN: phosphatase and tensin homolog deleted on chromosome ten, PLC: phospholipase C, PI3K: phosphoinositide-3 kinase, AKT also known as PKB: protein kinase B, GSK3: glycogen synthase kinase 3, RAS/RAF/MEK/ERK: mitogen-activated protein kinase signaling pathway, STAT5: Signal transducer and activator of transcription 5, mTOR: mammalian target of rapamycin).
The study of UCN-01 and midostaurin has shown that modifications at the 3’-N and C-7 positions can improve the drug formation of staurosporine. Early studies on halogenated staurosporine derivatives revealed that halogens at the C-3 position could increase the selectivity and activity against some tumor cells
[80][81][82][91,92,93]. This insight led to the synthesis of a series of staurosporine derivatives (
120–
143) (
Figure 8) with modifications at the 3′-N, 3-, and 7- positions.
Moreover, enzastaurin (
144) (
Figure 8 and
Figure 9) was initially studied as an isoenzyme-specific derivative of staurosporine, targeting and inhibiting PKC β, an activator of tumor development. Currently, it is in clinical phase III trials but faces several challenges. The progress in the research and development of the anticancer drug enzastaurin has been detailed in a review by Johnston, R.N
[83][96].