Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlos Sinogas | -- | 8832 | 2024-03-04 12:48:00 | | | |
| 2 | Camila Xu | Meta information modification | 8832 | 2024-03-05 02:19:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cordeiro, M.A.; Vitorino, C.; Sinogas, C.; Sousa, J.J. A Regulatory Perspective on Biosimilar Medicines. Encyclopedia. Available online: https://encyclopedia.pub/entry/55826 (accessed on 25 July 2026).
Cordeiro MA, Vitorino C, Sinogas C, Sousa JJ. A Regulatory Perspective on Biosimilar Medicines. Encyclopedia. Available at: https://encyclopedia.pub/entry/55826. Accessed July 25, 2026.
Cordeiro, Marta Agostinho, Carla Vitorino, Carlos Sinogas, João J. Sousa. "A Regulatory Perspective on Biosimilar Medicines" Encyclopedia, https://encyclopedia.pub/entry/55826 (accessed July 25, 2026).
Cordeiro, M.A., Vitorino, C., Sinogas, C., & Sousa, J.J. (2024, March 04). A Regulatory Perspective on Biosimilar Medicines. In Encyclopedia. https://encyclopedia.pub/entry/55826
Cordeiro, Marta Agostinho, et al. "A Regulatory Perspective on Biosimilar Medicines." Encyclopedia. Web. 04 March, 2024.
Copy Citation
By definition, biosimilar medicinal products are biological medicinal products that are similar to other biological medicinal products that are already on the market—the reference medicinal products.
biosimilar medicines
regulatory perspective
European Medicines Agency
1. Introduction
Biological medicinal products are defined as active substances obtained from a biological source, such as living cells or organisms, most of which are composed of proteins. Manufacturing these products involves complex biotechnological processes, setting them apart from those obtained through chemical means. Due to their intricate molecular structure and inherent variability, biological medicinal products undergo stringent quality and safety assessments [1][2]. These medicines typically contain one or more active substances sourced from biological materials. Reference biological medicines undergo a thorough evaluation based on comprehensive technical and scientific documentation covering aspects of quality, safety, and efficacy [3].
2. Definition and Characterisation of Biosimilars
According to the EMA, a biosimilar medicine is considered to be a medicine highly similar to another biological medicine that is already marketed in the EU—a reference medicine. Since biosimilars are a type of biological medicinal product, all relevant characteristics of biological medicinal products apply to them. Biosimilar medicinal products are molecules of high molecular weight with high complexity and are produced in cells or transgenic organisms [1].
A biosimilar medicine is developed to be very similar to its reference medicine in terms of quality, safety, and efficacy. Although there may be minor differences due to the complex nature and production methods, the active ingredients of a biosimilar medicine and its biological reference medicine are essentially the same biological substance [4].
In terms of similarity, the biosimilar medicinal product has physical, chemical, and biological properties highly similar to the reference medicine, particularly in terms of safety and efficacy. As for the existing variability, this may exist on a small scale, and it is crucial to demonstrate that it does not affect the safety and efficacy of a biosimilar. As for general standards, all biosimilar medicinal products comply with the same quality, safety, and efficacy standards as any other medicinal product [1]. However, being biological products, there is an associated variability that prevents the biosimilar medicine from being exactly equal to the reference medicine [5][6].
For the safety and efficacy profile to be maintained, legislation mandates the studies to be conducted so that a biosimilar medicine can demonstrate similarities in terms of quality, safety, and efficacy to its reference medicine and that there are no significant differences [7]. Table 1 shows the comparison between the data requirements for the approval of a biosimilar and a reference medicinal product [1].
Table 1. Data requirements for the approval of a biosimilar and a reference medicinal product. (Adapted from [1]).
| Reference Medicinal Product | Biosimilar Medicinal Product |
|---|---|
| Risk Management Plan | Risk Management Plan |
Clinical studies:
|
Comparative clinical studies:
|
| Non-clinical studies | Comparative non-clinical studies |
| Pharmaceutical quality studies | Comparative quality studies: Pharmaceutical quality studies, namely, full data requirements regarding pharmaceutical quality, as well as additional quality studies comparing the structure and biological activity of the biosimilar medicine with the reference medicine. |
It should also be noted that the clinical and non-clinical data required for the approval of a biosimilar are different from those required for the approval of a biological medicine with a new active substance. This is because the biosimilar relies on the experience gained with the reference medicine in terms of efficacy and safety to demonstrate similarity.
Biosimilar medicines are approved in an abbreviated procedure that guarantees similarity, but not equivalence, with the pre-existing reference medicine. In this context, comparability studies make it possible to generate the evidence required to substantiate the similarity in the areas of quality, safety, and efficacy, thus guaranteeing that the efficacy and safety identified for the reference medicine are recognised for the biosimilar medicine.
However, how is similarity addressed? How is the absence of significant clinical differences reported? High similarity and no significant clinical differences from the reference product are the two key requirements for biosimilar products. Recent reviews have addressed the pivotal regulatory considerations to support similarity assessment so that these questions are answered and clarified. In this sense, the assessment of similarity first involves a structural and functional characterisation/evaluation, followed by non-clinical and clinical studies, adopting an adapted approach throughout the biosimilar development process. It is important to emphasise that the aim of comparative clinical studies is not to re-establish efficacy and safety but to identify any differences considered clinically significant between the proposed biosimilar and the reference medicine. Nevertheless, the approval of biosimilar medicines is highly regulated, so differences between the biosimilar medicine and its reference medicine must be supported by strong scientific evidence that these differences are not clinically significant [8][9]. The World Health Organization also presents a guideline on the evaluation of biosimilars, in order to outline the globally accepted principles for licensing biosimilar medicines, i.e., medicines that are highly similar to reference biological medicines in terms of quality, safety, and efficacy [10].
3. Development of Biosimilar Medicines
In terms of development, biosimilars entail costs approximately one hundred times higher than those associated with generic drugs, with a development timeline ranging between seven and eight years, as opposed to the two to four years typically required for generic drugs [11]. The basis of the development of a biosimilar drug is thus an extensive structural and functional characterisation as well as the comparison of the biosimilar drug with the reference drug.
The development of a biosimilar medicine starts with defining the fingerprint of the reference medicine and its attributes, establishing the boundaries of potential variability for the biosimilar. Since the manufacturing process of the reference molecule remains undisclosed, a novel process must be devised to ensure alignment with the fingerprint. Such a process requires that the cell culture and purification process conditions are continuously adjusted, exploring new cell lines throughout the development until the highest similarity is achieved. Progress towards clinical trials for the potential biosimilar hinges on achieving full characterization of the molecule, well-defined processes, and confirmed molecular similarity [7][12][13]. The development process for biosimilars is complex, encompassing various stages such as cell line selection, cultivation, production, isolation, purification, and, ultimately, formulation, filling, and finishing.
The first decision in the development of a biosimilar is about the cell line creation (Figure 1), being such an important decision for the glycosylation patterns and the determination of the final profile of the biosimilar so that the protein of interest is expressed. The relevant gene is then cloned into a complementary DNA vector, which could be derived from human or microbial cell lines. Generally, the industry opts for Chinese hamster ovary cells for expression cells due to their consistent folding, ability to thrive in suspension, high yield, and stability against changes in pH and oxygen levels [14][15][16][17]. Following this step is clone selection, aiming to identify clones that closely mirror the desired product fingerprint. It is crucial to note that no two biologics are identical as each product is manufactured using a unique cell line from the producer [18]. It is important to mention that there are no two identical biologics because each product is produced using an exclusive cell line from the manufacturer [19][20].
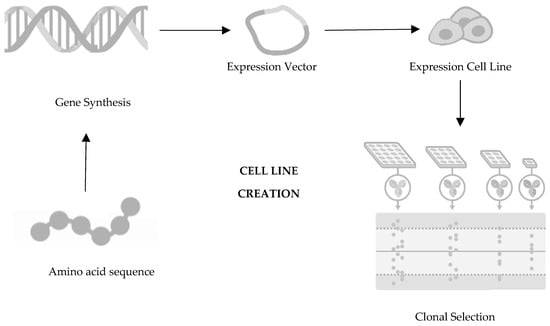
Figure 1. First step of development of a biosimilar: cell line creation. Adapted from [21].
Moving towards the second step in the development of a biosimilar, cultivation and production (Figure 2), the cell line that will produce the protein and originate the biosimilar medicine is expanded in a fermentation medium to obtain a batch of master cells, and these cells are then cultivated and grown in large-scale bioreactors [14][15][22][23]. First, thawing of the cells occurs, followed by inoculation under agitation to increase cell density. To ensure cell viability, the cells are maintained in a growth medium fortified with nutrients and supplements [24]. Finally, it is also important to mention that throughout this process, parameters such as the amount of oxygen, lactate production, temperature, pH, and osmolarity are controlled [20][25].
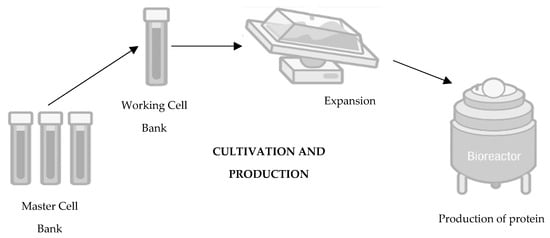
Figure 2. Second step of development of a biosimilar: cultivation and production. Adapted from [21].
The third step consists of isolation and purification (Figure 3). It is intended to recover the target protein, discarding unwanted impurities whether they are viruses, proteins, host cell DNA, aggregates, or endotoxins [26]. At this stage, the protein of the biosimilar is excreted in the cell culture fluid by the mammalian cell, and it is necessary to recover it by removing cellular debris by centrifugation and filtration. In addition, cellular metabolites are also eliminated so that they do not generate immunogenicity [24]. Still, within the scope of purification, it is important to highlight that such a process depends on column chromatography and filtration to remove unwanted impurities, using the biochemical properties that are similar to what is intended to be eliminated [20]. At this stage, the procedures are also constantly monitored, namely, pH, conductivity, and flow rate [20][27].
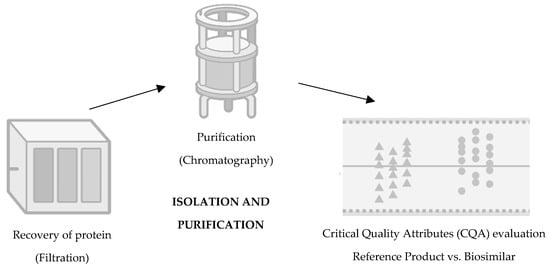
Figure 3. Third step of development of a biosimilar: isolation and purification. Adapted from [21].
Finally, the last stage concerns the following topics: formulation, fill, and finish (Figure 4). The formulation of the product inevitably influences the mimicking of its degradation and the maximisation of its shelf life, involving the optimisation of the buffer solution conditions, such as pH, ionic strength, excipients, and stabilisers [24]. In this stage, the product is subject to specific conditions to assess its behaviour and stability, such as exposure to high levels of agitation or thermodynamic stress, such as high temperatures, freezing, or thawing for long periods. Regarding the final presentation of the drug, it can take three possible forms: liquid, frozen liquid, or lyophilized. The preferred option is typically the liquid form, although this choice depends on the stability of the product [7][20]. Freezing the product during storage may be necessary to mitigate chemical degradation. Opting for a lyophilized presentation entails additional costs and necessitates the use of diluents for reconstitution. Alternatively, if the frozen form is chosen, a cryoprotectant such as sucrose should be added to minimize cryoprecipitation or aggregation caused by cryoconcentration [20][25].
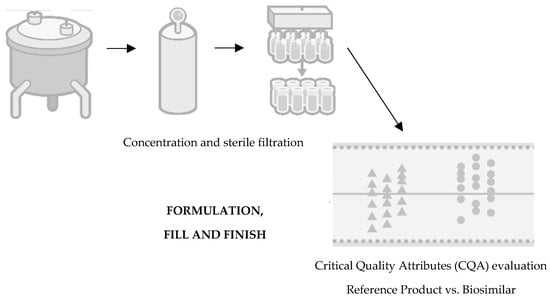
Figure 4. Fourth step of development of a biosimilar: formulation, fill, and finish. Adapted from [21].
The manufacturer of the biosimilar medicine must develop the entire manufacturing process, from cell line selection to production. These activities are performed without full access to the development history of the reference medicine. Consequently, there exists significant potential for variations between an innovative biological medicine and a biosimilar medicine in terms of manufacturing processes. Therefore, any differences in the safety/efficacy profile should always be justified. It should also be noted that modifications for the purpose of improving efficacy are not expected to be accepted as distinct from the concept of a biobetter. The comparative studies between the biosimilar biological medicine and the reference medicine include the criteria of quality, safety, and efficacy, all aimed at demonstrating compliance with approved regulations [28].
Pharmaceutical quality then focuses on characterisation at the structural level, through physicochemical properties, which encompasses control and production standards and product preparation and processing. In addition, purity must also be controlled so that it does not exceed the established limit. Other data concern the biological activity, the excipients and starting materials, the dosage and formulation, the control of the manufacturing process, and the actual stability of the active substance and the final product [1].
3.1. Immunogenicity
Concerning immunogenicity, proteins inherently possess the capacity to elicit an undesired immune response. In rare instances, this reaction can lead to severe adverse effects or even diminish efficacy, although such occurrences are infrequent. Immunogenicity is influenced by various factors, including the drug characteristics, external factors associated with treatment, and individual patient- or disease-related factors. Quality issues, such as alterations in production methods, formulations, or packaging, can impact immunogenicity by affecting the drug properties and impurity profile. However, it is crucial to ensure that these changes do not compromise the medicine’s efficacy and safety [29][30]. It is very unlikely for an adverse immune reaction to occur following the aforementioned modifications as comparability studies indicate no significant increase in impurities or aggregates despite these alterations. Regulatory agencies actively monitor the immunogenicity of biosimilar drugs, employing comparability studies across different batches and utilizing physicochemical and structural analyses alongside functional in vitro assays [31][32].
The immunogenicity data required for the approval of a biological medicinal product for approval purposes, as highlighted by the EMA, include the incidence, titration, and persistence of antibodies against the biological medicinal product; neutralisation tests; clinical impact assessment; and measures to manage the risk of immunogenic potential. Nevertheless, these data always depend on the type of biological medicine and its intended use and the characteristics of the product itself, among others [1][33].
3.2. Extrapolation
Extrapolation represents a well-established scientific concept, aiming to evaluate the extent to which a biosimilar closely resembles a reference medicinal product in terms of safety and efficacy within a particular therapeutic indication. This similarity allows for the extension of these findings to other approved indications for the reference product. In practical terms, this means that, in some situations, few clinical studies with biosimilars may be carried out since this extrapolation is always supported by scientific evidence from comparability studies. Extrapolation criteria typically include considerations such as the mechanism of action, the target study population, various clinical settings, safety profiles, and immunogenicity data [1][34].
Concerning the mechanism of action of the active substance, it should be mediated by the same receptor, and additional research may be necessary to demonstrate similarity in behaviour. Depending on the clinical context, variations in dosage, pharmacokinetics, or mode of action may arise, necessitating further studies as the data for one indication may not directly apply to another. Establishing a comparable safety profile for a specific indication is crucial at this stage before extrapolating safety data. Immunogenicity data, being a specific criterion, require thorough substantiation and add complexity to the process [1][35].
3.3. Safety
In addressing the safety of biosimilar medicinal products, a comprehensive approach is essential, from a sound regulatory framework, a risk management plan always in place, post-authorisation safety studies, and continuous safety monitoring. Given that many adverse drug reactions associated with biosimilars can be anticipated based on their pharmacological effects, efforts are made to collect reports of adverse reactions spontaneously. This includes the submission of periodic safety reports and the implementation of additional monitoring mechanisms to detect potential long-term adverse events or with long latency periods [1].
The EU has a consolidated system of monitoring, reporting, assessing, and preventing adverse drug reactions, so the authorities continuously assess the risk–benefit ratio of all medicines and take the necessary measures. When an undertaking applies for the marketing authorisation, it will submit a risk management plan including a pharmacovigilance plan as well as any risk minimisation measures to identify, characterise, and minimise risks. When talking about a risk management plan (RMP) for a biosimilar medicine, it will always be based on the knowledge and experience already gained with the reference medicine. For post-authorisation studies, it has been made possible to monitor known risks and detect adverse drug reactions that only arise after a large number of patients receive treatment over a substantial time period. In addition, it is noteworthy that toxicity studies are used to carry out this safety assessment of biological medicinal products. The main mechanisms of adverse effects of biological drugs are related to exaggerated pharmacology. This phenomenon, known as on-target toxicity, entails the occurrence of adverse pharmacological effects specifically at the intended target site. However, if comparable pharmacological activity has been established in vitro, there is no need to confirm these mechanistic properties in vivo [36].
Examples of unexpected toxicity are scarce. Often, functional differences between these antibodies have a structural basis rooted in variations in the amino acid sequence. These differences are not integrated into the development of similar biological medicines because the amino acid sequence should be the same as that of the reference medicine [37]. Thus, although unexpected toxicity can be found in pre-clinical animal studies during the development of new biological medicinal products, there is no evidence to suggest that such occurrences are associated with biosimilars [12].
3.4. Comparability
The fundamental principle of developing a biosimilar is based on the comparability exercise that is carried out between the reference biological medicine and the biosimilar medicine. For such a comparability exercise to be undertaken, comprehensive analyses of the proposed biosimilar and the reference product are required, using sensitive methods. The main objective is to demonstrate that the biosimilar and the reference product are similar at a finished medicinal product level [28].
In this context, several important pillars arise when comparing the reference medicine and the biosimilar medicine: target/quality by clinical trial design, quality (data set for stand-alone) biological and physicochemical comparability, comparative pre-clinical trials, comparative clinical trials, and risk management plan [11]. The comparability exercise is carried out in several steps, namely, quality comparability, non-clinical comparability, and clinical comparability.
In the first step, quality comparability, a full characterisation approach needs to be undertaken to compare the physicochemical and biological quality attributes, for instance, the purity of the potential biosimilar medicine. This is performed using a wide range of different state-of-the-art analytical tests, as no single method can characterise all aspects of a product. The development process is also to be modified if there are significant differences found in the analyses until the product generated has a profile that matches the reference product profile. Iterative adjustments are made at every phase of the development process to ensure that the ultimate biosimilar medicine meets the quality standards set by the EMA, aligning with all criteria stipulated for documentation submission, assessment, and marketing authorization.
In the second step, non-clinical comparability, non-clinical studies, sometimes called pre-clinical studies, need to be conducted for biosimilar medicines before any clinical trial begins. Generally, non-clinical data for a biosimilar are generated through an abbreviated testing programme or in vivo animal studies, as required by the EMA’s guidelines. On the other hand, non-clinical studies usually include repeated dose toxicity studies as well as pharmacokinetic and pharmacodynamic (PK/PD) studies along with local tolerance testing. For this type of study, similarity criteria should be pre-defined and scientifically justified in order to allow comparability of the support with the reference and detect potential differences between them.
In the third step, clinical comparability, clinical tests are also considered comparative in the case of the development of a biosimilar medicinal product. However, such clinical tests are not required to the same extent as would be necessary for a new active substance, taking into account the clinical experience gained from the use of the reference medicinal product. Therefore, it is crucial to consider the nature and characteristics of the medicinal product as well as the therapeutic indications. Another key aspect is to understand how comparable the profile of the biosimilar is to that of the reference product. The closer the biosimilar and reference profiles are and the greater the similarity demonstrated (through appropriate studies such as comparative quality, biological and receptor binding activity analyses, and animal testing), the more easily the clinical trial programme will be accepted by the regulatory authorities.
In summary, the clinical comparability exercise starts with pharmacokinetic and/or pharmacodynamic studies, and comparative clinical efficacy and safety trials may still follow. An important step is to document the side effects and to take into consideration the evaluation of immunogenicity for which comparable profiles for the biosimilar and the reference are also required for the clinical safety data assessment [38][39]. Apart from comparing the quality attributes of both medications, establishing the similarity in biological activity and safety of the biosimilar involves utilizing relevant and sensitive in vitro assays. These assays help identify significant differences in allosteric mechanisms between the two medications across all approved indications [40]. Table 2 outlines the comparability critical attributes and the way the similarity of the products is demonstrated [12][40].
| Comparability Critical Attributes | Similarity Demonstration |
|---|---|
| Protein structure and production quality | Extensive laboratory analysis for all molecular characteristics (multiple lots) |
| Pharmacokinetics, Pharmacodynamics, and Animal Toxicity | In vitro and in vivo assays (carried out on relevant animal species, if further confirmation scans are required for laboratory studies) |
| Pharmacokinetics, Pharmacodynamics, and Animal Toxicity | Phase I clinical studies |
| Clinical Efficacy and Safety | Phase III clinical studies |
| Safety in clinical practice | Risk management plan Phase IV studies Pharmacovigilance |
In terms of protein structure, as a critical quality attribute, the similarity assessment includes a detailed analysis of conformational and post-translational modifications between a biosimilar and its reference medicine. This is fundamental in demonstrating comparability and ensuring the efficacy and safety of the biosimilar. These structural characterization studies involve sophisticated analytical techniques such as mass spectrometry, nuclear magnetic resonance spectroscopy, and X-ray crystallography to provide insights into protein folding, stability, and function. A thorough understanding of protein structure similarity is critical in establishing the interchangeability and substitutability of biosimilar medicines, ensuring patient safety and therapeutic efficacy [41].
3.5. Interchangeability
The term interchangeability emerged in the context of the possibility of exchanging a reference biological medicine for its biosimilar, or a biosimilar for another biosimilar, hoping to obtain the same therapeutic effect produced by the option initially used [1][42].
This exchange can occur in two distinct ways: switch or automatic substitution. As for the “switch” option, this occurs when the prescribing physician decides to exchange one medicine for another that has the same therapeutic purpose. Automatic substitution, on the other hand, refers to a practice carried out at the pharmacy level without consulting the prescribing physician, where the dispensing of the prescribed drug is changed by another equivalent and interchangeable drug [1][43].
These issues have been the subject of some controversy, despite the existence of several studies that prove the safety and effectiveness of this practice [44].
In terms of pharmacovigilance, the traceability of the biological medicine involved in the potential adverse reaction is very important, so the same brand of medicine should be kept for as long as the reaction is expected to occur. The change between biosimilar biological medicines should respect a minimum period of time that safeguards their traceability. This information can be found in the National Medicines Formulary and, when this is not mentioned, that period should not be less than 6 months. Switching between different brands of the same biological medicine must be articulated with the clinical services involved, with respect to the precautionary principle and in accordance with the therapeutic indications for each situation. This position is reviewed whenever applicable scientific evidence becomes available.
Therefore, what is the EMA’s position on the issue of switching the biosimilar as a reference medicine? Previously, the EMA positioning was directed to the independent deliberation by each member state.
In Portugal, the decision regarding therapeutic switching is made by INFARMED I.P. in conjunction with the National Pharmacy and Therapeutics Commission. They stipulate that the switch should occur only after the specified minimum duration to guarantee medicine traceability. However, automatic substitution is not currently permitted in Portugal [1][45][46]. Note, however, that in September 2022, as reported on the EMA’s website, the EMA and the Heads of Medicines Agencies (HMA) released a joint statement affirming the interchangeability of biosimilar medicines approved in the EU with their reference medicine or an equivalent biosimilar. This unified stance serves to standardize the approach across the EU [47].
4. Regulatory Framework for the Development of Biosimilars
The marketing authorisation for biosimilar medicinal products, via the EMA, is obtained through a centralised procedure, in which the Agency evaluates the medicinal products for the purpose of authorising their marketing. Comparative characteristics studies evaluate the composition, physical properties, protein structure, purity, isoforms, or impurities that derive from the product, as well as biological activity. The production process between the reference biological drug and the biosimilar may, however, present differences. Concerning the regulatory framework for the development of biosimilars, the perspective of the approval process via the EMA and FDA as well as other regulatory agencies may differ in certain aspects [6][48].
4.1. European Medicines Agency
All biotechnologically produced medicinal products are approved in the EU by the EMA through a centralised procedure. There are certainly some biosimilars, such as low-molecular-weight heparins obtained from the swine intestinal mucosa, which are approved at the national level; however, mostly, and since they resort to biotechnology for production, they are approved by the centralized procedure.
First, the entity submits the marketing authorisation application to the EMA. The data are then evaluated by the scientific committees, the Committee for Medicinal Products for Human Use (CHMP) and the Pharmacovigilance Risk Assessment Committee (PRAC), as well as by experts in biological medicinal products and biosimilar specialists. Subsequently, this analysis gives rise to a scientific opinion sent to the European Commission (EC). If applicable, it is for the EC to grant marketing authorisation in the EU and it is valid in all EU member states [1].
The EMA was the first organisation to delineate a specific regulatory route for biosimilar medicines; this happened in October 2005. The EMA guidelines state that the approval of biosimilars is based on comparability studies, through the characterisation of the protein structure of the biosimilar, as well as its efficacy, safety, and immunogenicity. In addition to these aspects, it is considered essential to carry out in vitro tests, impurity profile analyses, and pharmacokinetic and pharmacodynamic studies, as well as pharmacovigilance monitoring after its approval on the market [49].
There are several important requirements according to the Agency, including the importance of demonstrating robustness and consistency in its manufacturing process, as well as details for additional non-clinical and clinical studies to attest to the comparability.
A key point highlighted by the EMA is that the clinical benefit has already been proven by the innovative product; so, the goal of a biosimilar is to demonstrate similarity to the innovative product, in terms of strength, active substance, route of administration, and pharmaceutical form, and not the clinical benefit itself.
In order to compile all legislation concerning biological medicines, the EMA has adopted several more individualised guidelines for each type of biosimilar medicine, as presented in Table 3. The general guidelines include the Guideline on similar biological medicinal products, the Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues, and the Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: clinical and non-clinical issues [4][28][31][50][51]. In addition, the EMA has also developed more specific guidelines: the Guideline on biosimilar medicines containing recombinant granulocyte colony-stimulating factor, Guideline on clinical and non-clinical development of similar medicines containing low-molecular-weight heparins, Guideline on clinical and non-clinical development of similar medicines containing recombinant human insulin and insulin analogue, Guideline on clinical and non-clinical development of similar medicines containing beta interferon, Guideline on clinical and non-clinical development of similar medicines containing monoclonal antibodies: ethical and non-clinical issues, Guideline on clinical and non-clinical development of similar medicines containing recombinant erythropoietin, Guideline on clinical and non-clinical development of similar medicines containing recombinant follicle-stimulating hormone, and Guideline on clinical and non-clinical development of similar medicines containing somatropin [4][28][31][50][51]. Finally, two other relevant guidelines on the comparability of biotechnology-derived medicinal products after a change in the manufacturing process exist: clinical and non-clinical issues and another one on ICH Q5E biological/biotechnology products subject to changes in their manufacturing processes: comparability of biological/biotechnology products [4][28][31][50][51].
Table 3. European guidelines for the development and evaluation of biosimilar medicines [52].
| Type of Guideline | Guideline Title |
| General | Guideline on similar biological medicinal products. |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues. | |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: clinical and non-clinical issues. | |
| Specific | Guideline on biosimilar medicines containing recombinant granulocyte colony-stimulating factor. |
| Guideline on clinical and non-clinical development of similar medicines containing low-molecular-weight heparins. | |
| Guideline on clinical and non-clinical development of similar medicines containing recombinant human insulin and insulin analogue. | |
| Guideline on clinical and non-clinical development of similar medicines containing beta interferon. | |
| Guideline on clinical and non-clinical development of similar medicines containing monoclonal antibodies: ethical and non-clinical issues. | |
| Guideline on clinical and non-clinical development of similar medicines containing recombinant erythropoietin. | |
| Guideline on clinical and non-clinical development of similar medicines containing the recombinant follicle-stimulating hormone. | |
| Guideline on clinical and non-clinical development of similar medicines containing somatropin. | |
| Others | Comparability of biotechnology-derived medicinal products after a change in manufacturing process: clinical and non-clinical issues. |
| ICH Q5E biological/biotechnology products subject to changes in their manufacturing processes: comparability of biological/biotechnology products. |
4.2. Food and Drug Administration
In the United States, the creation of guidelines for biosimilars did not happen as early as in the EU. The FDA has only recently started formulating distinct guidelines for the approval of biosimilar drugs. At the level of important factors to consider at the time of the FDA’s approval process, the need for robustness in the manufacturing process, the similarity of the protein structure, the extent of understanding of the mechanism of action, appropriate pharmacodynamics trials, comparative pharmacokinetic data, immunogenicity data, and clinical data regarding the innovative product are highlighted [49].
In the initial phase, the biosimilar medicinal product entity presents a series of data comparing the candidate medicinal product with the reference medicinal product in order to demonstrate similarity. First, the data are evaluated, starting with a detailed analytical characterisation, at the structural and functional level, as well as a comparison, moving to animal studies, and, if necessary, comparative clinical studies [18].
The submission of a biosimilar medicinal product will include some data to demonstrate such similarity with the reference medicinal product, from analytical studies demonstrating that the biosimilar medicinal product is highly similar to the reference medicinal product to toxicity assessment studies. Another aspect refers to a clinical study to demonstrate the safety, purity, and potency of the proposed biosimilar product in one or more of the indications for which the reference product is licensed. This typically includes the evaluation of immunogenicity, pharmacokinetics (PK), and, in some cases, pharmacodynamics (PD) and may also include a comparative clinical study [53].
More practically, the application for marketing authorisation is made to the FDA and submitted through section 351(k) of the Public Health Service Act (PHS). This section allows the biosimilar medicinal product to be approved for all therapeutic indications of the reference medicinal product. This happens several times when discussing the speed of the drug approval process in the United States. This situation is due to the fact that, in 2010, a change was made to the Biologics Price Competition and Innovation Act, enabling the existence of a shortened route for the regulation of biosimilar drugs in the USA. However, it is important to point out that such a change does not diminish the required standards of safety, purity, and efficacy.
As with the EMA, the FDA requires a rigid, sequential approach with rigorous comparability tests, from physical-chemical, analytical, functional, and non-clinical and clinical assessments.
In 2014, the Purple Book was published by the FDA, where reference drugs and corresponding biosimilars are found, presenting the medicines authorised for commercialisation in the USA [54][55]. In 2015, the FDA published three guidelines, granting industry guidelines for the development of biosimilar drugs. These include scientific assessments in the demonstration of similarity, considerations about the quality for the demonstration of similarity with the reference medicinal product, and a set of questions and answers on the evaluation of the drugs in question [16]. In 2018, the FDA’s Biosimilar Action Plan (BAP) was published, providing information regarding actions to stimulate the development of biosimilars. Briefly, the BAP analyses the phases so that development and approval are more effective, clarifying regulation and developing support materials for healthcare professionals and patients to demystify the issue and increase confidence. Nevertheless, the BAP does not address issues related to improvements in pharmacovigilance [54][56].
Table 4 summarises the key criteria for developing a biosimilar medicine through FDA legislation. The FDA’s determination of similarity is based on the totality of evidence provided in the marketing application for FDA review. The data set in the marketing application includes extensive analytical comparison to show that the proposed biosimilar and the reference products are extremely similar in structure and function. Animal, human pharmacological, immunological, and other data are added as needed to the analytical data in a stepwise manner to provide the necessary information with the ultimate goal of demonstrating similarity [13].
Table 4. FDA’s development of a biosimilar medicine. Adapted from [13].
| FDA advice on the scope and extent of testing during development | Highly Similar | The totality of the evidence | |
| Analytical Studies | Quality characteristics assessment, using state-of-the-art technologies and multiple different tests for the same characteristic, to determine whether the proposed biosimilar is highly similar to the reference product. Identification of differences in quality characteristics, if applicable, between the reference product and the proposed biosimilar (examples of quality characteristics may include structure and bioactivity). Evaluation of the potential impact of any observed differences. |
||
| Toxicity’s Assessment | |||
| Animal Studies | Support safety decision making before human exposure to the proposed biosimilar. Additional support to demonstrate similarity (but not always necessary). |
||
| No Clinically Meaningful Differences | |||
| Human PK and PD Studies | Comparison between the pharmacokinetics (exposure) and, if applicable, the pharmacodynamic (response) profiles of the proposed reference and biosimilar product to support a conclusion of similar efficacy and safety. Human PK and PD studies are generally considered to be the most sensitive data element to support a demonstration of no clinically meaningful differences. |
||
| Immunogenicity Assessment |
Comparison between the incidence and severity of immune responses generated with the reference product and the proposed biosimilar. Immunogenicity assessment is generally included as part of all clinical studies. |
||
| Additional Clinical Studies | Additional clinical studies when residual uncertainties remain about demonstrating that there are no clinically meaningful differences after conducting the above-mentioned studies. | ||
| Experience with the Reference Medicine | |||
| Biosimilarity: According to the general criteria defined by the FDA, the demonstration of biosimilarity is based on the totality of evidence provided when submitting a marketing application for FDA review. The presented data include an extensive analytical comparison in order to show that the proposed biosimilar medicine and the respective reference product are extremely similar in terms of structure and function. Moreover, animal, human pharmacological, immunological, and clinical data are enhanced as necessary to the analytical data, in a stepwise approach to provide the required information to demonstrate biosimilarity. | |||
4.3. Other Regulatory Authorities
In addition to the EMA and FDA, there are other regulatory agencies also responsible for the evaluation and monitoring of medicines submitted for marketing authorisation.
In Japan, the Pharmaceuticals and Medical Devices Agency (PMDA), in particular, the ‘Office of Cellular and Tissue-Based Products’, stands out as the organisation responsible for the assessment, regulation, and authorisation of biosimilar medicines, dealing with a wide range of activities, from approval reviews to post-authorisation surveillance. The Japanese regulation is based on European legislation and, therefore, bears a strong resemblance to the binding model of the EMA. The PMDA also highlights the need to conduct clinical trials in order to demonstrate clinical comparability between the biosimilar medicine and the reference medicine. Nevertheless, sometimes conducting comparative studies on pharmacokinetics and pharmacodynamics can be considered sufficient to demonstrate clinical comparability and no further clinical trials are required [57][58]. Another debated point from Japan concerns extrapolation, and this can be carried out for all therapeutic indications of the reference medicinal product, provided that equivalent therapeutic effects and clinical comparability are demonstrated [59][60].
Regarding the Middle East market, the emergence of biosimilar medicines has allowed such medicines to reach other countries characterised by a high percentage of poverty, avoiding the poor-quality imitations that are so typical of these regions [35]. The Central Drugs Standard Control Organisation is the organisation responsible for medicines in India, and its mission is to ensure that all procedures are reliably controlled. Under Indian law, a biosimilar medicine can be approved faster if its reference medicine is approved in the EU or the USA. For example, a biosimilar can be approved with phase III bioequivalence studies with about 100 patients, while the EMA requires much more, up to 500. A major peculiarity of the Indian legislation is the lack of market exclusivity for products that are approved for the first time. In these cases, this exclusivity relates only to the patent plus the fact that the patent is less developed and, therefore, considered a lower barrier [59][61].
In Canada, the drug approval process is handled by the Biologics and Genetic Therapies Directorate (BGTD) of the Health Products and Food Branch (HPFB) of Health Canada. Currently, there are specific guidelines for biosimilar medicines prepared by Health Canada to have more authoritative legislation. The first biosimilar drug approved in Canada was a growth hormone, through Sandoz, called OMNITROPE, in 2009 [62][63].
In South America, regulatory guidelines follow the EMA’s and FDA’s models, with some particularities certainly, as will be shown in the following paragraphs.
For the registration of biosimilars in Venezuela, the National Institute of Hygiene Rafael Rangel is used; it is based on the generation of information on biosimilarity in terms of the quality, safety, and efficacy of the reference medicine. It should be noted that extrapolation to use indications is not allowed, so individualised/separate information is generated [48][64].
The Instituto Nacional de Vigilancia de Medicamentos y Alimentos is the regulatory authority in Colombia and it is responsible for the inspection and supervision of the marketing and development of health products, as well as the identification and evaluation of sanitary standards and procedures, among others [46].
In Brazil, according to Brazilian legislation and the National Health Surveillance Agency, the registration of biosimilars is performed after the submission of comparative studies between the biosimilar and the reference medicine that confirm the quality, efficacy, and safety requirements. When a biosimilar drug is in the approval stage, extensive pre-clinical documentation is required, which should show such points of similarity, ensuring its criteria and attributes [45][65].
In Chile, the responsible regulatory agency is el Departamento Agencia Nacional de Medicamentos, and, concerning biosimilars, this is still a developing country in terms of the regulation necessary for them [49].
Argentina’s regulatory agency is called la Administración Nacional de Medicamentos, Alimentos y Tecnología Medica and it is governed by the guidelines of the EU model [66].
In China, the Center for Drug and Food Evaluation of the China Drug Administration issued guidelines on “similar biologics” on 28 February 2015. According to the Chinese guidelines, a similar biologic is a “therapeutic biologic” similar to an authorised reference product in terms of quality, safety, and efficacy. Regarding clinical trials, the Chinese guidelines suggest that clinical trials for “similar biologics” should start with PK and PD studies based on which clinical efficacy and safety studies should be conducted. Comparability between the similar biological product and the reference product may be performed for quality assessment and non-clinical studies based on PK, PD, and PK/PD clinical studies. The Chinese guidelines suggest that the assessment of clinical immunogenicity should be based on the results of a non-clinical immunogenicity assessment. Where such studies suggest the similarity of the similar biological product to the reference product, then only limited clinical evaluation is required. The approval of “similar biological products” has not been assigned to a separate abbreviated approval route. The approval pathways are subject to the same pathways that apply to innovative biological products. In addition, for the reference product, the Chinese authorities require the product to be approved in China. Reference products approved by foreign regulatory authorities and not approved in China are not acceptable as reference products [53][54].
The Ministry of Food and Drug Safety (MFDS) of the government of South Korea, through its National Institute for Food and Drug Safety Evaluation, seeks to scientifically evaluate drugs developed by South Korea’s pharmaceutical industries. Of note, the national guidelines were based on those of the World Health Organization, the EU, and Japan [52].
Some countries in Asia do not have their own agency that focuses on the evaluation of biosimilar medicines. Hence, the Association of Southeast Asian Nations (ASEAN) emerged; it represents 10 member states in the ASEAN region including Indonesia, Malaysia, Thailand, the Philippines, Singapore, Darussalam, Vietnam, Laos, Myanmar, and Cambodia. In this sense, it is through this Association that many decisions of scientific, political, and economic issues, among others, are made in order to have an important role in this region [67].
5. Pharmaceutical Market for Biosimilars
Based on a market research report, the biosimilar medicines market demonstrates continuous growth. This trend persists as numerous biological molecules enter the market, introducing novel therapeutic options across various therapeutic areas for patients [68].
5.1. Remicade®, a Reference Medicine
In order to understand the regulatory framework of a biosimilar medicine, a more practical study of Remicade® and its biosimilars was undertaken. The approval process described was centralised by the EMA.
Remicade® is an anti-inflammatory drug, usually used when other medicines or treatments have failed in adults with the following diseases: rheumatoid arthritis, Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and psoriasis [69].
In the case of rheumatoid arthritis, it is an immune system disease that causes inflammation of the joints, and Remicade® is given together with another medicine, Methodrex. Crohn’s disease is a disease of inflammation of the digestive tract, which can lead to the formation of fistulas. Ulcerative colitis and ankylosing spondylitis are diseases that cause inflammation, under the effect of ulcers in the lining of the intestine and pain in the joints of the spine, respectively. Finally, psoriatic arthritis and psoriasis cause the appearance of desquamative red plaques on the skin, the first of which simultaneously causes inflammation of the joints.
Regarding how Remicade® is used, this is the preparation of a solution for infusion in the form of powder. At the time of treatment, all patients are monitored for possible reactions, and it is therefore strictly crucial that it be administered with the supervision of a specialist doctor.
Remicade® may also be used in younger patients, aged 6 to 17 years, who have Crohn’s disease or severe ulcerative colitis and who have not been responsive to other medicinal products/treatments or even are unable to receive such therapeutic strategies [69][70]. Nevertheless, there are few biosimilars with therapeutic indications approved for paediatrics. However, the administration processes are more invasive, using the parenteral route, which means an added difficulty for children. In this sense, this point is still considered a delicate issue, and the regulatory agencies themselves do not always agree in this area [71][72].
The active substance in Remicade® is infliximab, which is a monoclonal antibody, a type of protein that is designed to recognize and bind to a specific structure antigen [69].
In practical terms, infliximab binds to a chemical messenger, tumour necrosis factor-alpha, and, once it is involved in the inflammation process, acts directly in this direction. Thus, it will promote TNF-α blockade, reducing inflammation and other symptoms associated with the disease in question [73][74].
Remicade® is a reference medicine with the following characteristics: 100 mg concentrate powder for infusion solution. Each vial contains 100 mg of infliximab. The mode of administration is intravenously over 2 h. Remicade® belongs to the pharmacotherapeutic group immunosuppressants, as inhibitors of tumour necrosis factor-alpha (TNF-α) [75].
Regarding its mechanism of action, the active substance (infliximab) is a man-murine chimeric monoclonal antibody that binds with a high affinity to both soluble and transmembrane forms of TNF-α but not a lymphotoxin (TNF-β). When administered at the recommended doses and concentrations, it is an effective, safe, and generally well-tolerated drug. Its mechanism of action involves the neutralization of the biological activity of TNF-α through the binding with high affinity to the soluble and transmembrane forms of TNF-α and inhibits the binding of TNF-α with its receptors. This neutralization leads to an overall reduction in inflammation [74].
5.2. Case Study: A Reference Medicine (Remicade®) vs. a Biosimilar Medicine (Inflectra)
The Remicade® approval process, as described in Figure 5, began on 27 March 1998, and the first evaluation report was released to all members of the Committee for Medicinal Products for Human Use (CHMP) on 8 June 1998. Subsequently, some questions were raised, and a consolidated list of questions was transmitted to the company on 30 November 1998. The evaluation report with the answers to these questions was then released by the rapporteur within the CHMP on 25 January 1999. On 24 February 1999, the CHMP discussed this same report and convened a group of experts to reflect on some specific clinical points, including safety, post-marketing, and efficacy studies. In this context, a list of quality aspects to be taken into account in this medicinal product was also recommended. Finally, on 19 May 1999, the CHMP, based on the data submitted as well as the scientific discussion within the Commission, issued a positive opinion for the granting of a marketing authorisation to Remicade®. It should also be reported that such opinions were also forwarded to the EC to follow up on this decision [76].

Figure 5. Approval process for Remicade® and Inflectra, via EMA’s centralised pathway [77].
Concerning the reference medicine Remicade®, four biosimilar medicinal products were approved for use in the European Area. The four biosimilar drugs (Inflectra, Remsima, Flixabi, Zessly) are anti-inflammatory drugs whose active substance is infliximab. In this sense, the four medicinal products are similar to a biological medicine, called a reference medicine, already authorised in the EU: Remicade®.
Such medications are used in the treatment of adults with the following diseases: rheumatoid arthritis, Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and psoriasis. Furthermore, they can be used in children between 5 and 17 years of age who have Crohn’s disease or severe ulcerative colitis and have been first submitted to other treatments or medications without success. Its mode of administration is via infusion form, into a vein for 2 h. With regard to the approval process, it has already differed among the four medicinal products, although it is similar [77][78][79][80].
5.2.1. A European Approach to Biosimilars’ Approval Process
Regarding the approval process of INFLECTRA, through the EMA, this was through a centralized procedure, as well as the others mentioned previously.
The procedure began on 14 July 2012, followed by a meeting between 16, as fully described in Figure 5, and 19 July 2012, in which the CHMP drew up a consolidated list of questions to send to the applicant, which was sent the following day to the applicant. On 16 November, the applicant submitted the answers to the consolidated list of questions. Following this, the rapporteurs released the joint evaluation report on such responses to all CHMP members on 9 January 2013. During the meeting from 7 to 10 January 2013, the Pharmacovigilance Risk Assessment Committee (PRAC) adopted the PRAC Council’s Risk Management Plan. Still within the CHMP, a list of outstanding issues was issued in writing or orally. On 29 April 2013, the applicant then submitted the answers to outstanding questions. To this end, the PRAC again adopted the Council’s RMP, and the rapporteurs released the joint evaluation report on such responses to all CHMP members on 24 May 2013. The applicant then orally stated the outstanding issues raised, between 27 and 30 May, and the CHMP agreed to a second list of outstanding issues to be addressed in writing by the applicant. After the applicant submitted the replies to the second list and the PRAC had gathered, the rapporteurs reissued the joint evaluation report and, on 27 June 2013, the CHMP, taking into account all the data collected and the scientific discussion generated, issued a positive opinion to grant a marketing authorisation. Figure 5 shows the approval process for Remicade® and its biosimilar Inflectra, via the EMA’s centralised pathway [77].
5.2.2. A United States of America Approach to Biosimilars’ Approval Process
It is also important to mention that Inflectra was the second biosimilar approved by the FDA and the first Remicade® biosimilar also approved for marketing in the United States. According to the FDA, the approval of Inflectra was based on a review of evidence including structural and functional characterisation, animal study data, pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and efficacy data that demonstrated that Inflectra is biosimilar to Remicade® [77].
6. A Critical Appraisal of Biosimilars
Reflection on biosimilar medicines becomes fundamental to bringing together ideas, discussing issues, and, essentially, reflecting on their benefits, risks, regulatory particularities, and future perspectives.
6.1. Challenges on Biosimilar Medicines
Currently, at the scientific level, one of the great challenges of efficacy and safety of biosimilar medicines lies in the difficulty of demystifying the concepts of biosimilar, generic, and biobetter. The three designations differ since generic medicines are equivalent to reference medicines and biosimilar medicines are highly similar to existing biological medicines. Regarding biobetter medicines, while the main objective of developing a biosimilar medicine is to clearly and evidently demonstrate high similarity to the reference medicine, the objective of a biobetter medicine is not to show similarity but to reveal beneficial clinical superiority for the patient over the original biological medicine. The studies already carried out and the knowledge about these medicines make it possible to educate society in this regard and make it more receptive to this development in science. It is therefore important to communicate, inform, and clarify to both patients and healthcare professionals that these therapeutic strategies are available at a lower cost but with the same quality, efficacy, and safety. The biggest difficulty/challenge in researching a biosimilar will always be to prove similarity and not non-inferiority to the reference drug.
Another challenge is the misinformation regarding these medicines, which promotes ignorance and naturally generates distrust regarding the quality, safety, and efficacy of biosimilar medicines. Nowadays, in the field of research, it is possible to state that during the whole process of development and production of a biosimilar, the quality, safety, and efficacy profiles are proven through clinical trials that assess the pharmacokinetics, efficacy, safety, and immunogenicity of the biosimilar compared to the reference one. In this way, it is proven, through tests that demonstrate that the properties of the product, which may affect its immunogenicity, have not been altered.
Bearing in mind that, although the market for biosimilar medicines is very complex, specific, and large, it also entails high costs. The cost required to develop a biosimilar medicine, in itself, is a challenge, considering the high-tech equipment and manufacturing material needed, making it very expensive throughout the process, including all the clinical trials needed to prove its efficacy, also highlighting the importance of investment in human resources, since it is essential to involve specialized personnel in different fields of science to ensure the proper development of this type of medicine.
One of the most referred to and extremely important challenges in the use of biosimilars refers to regulation, which allows professionals to have safety and trust in their administration. In general, the existing legislation can still be considered quite recent, causing a certain instability and insecurity in its implementation in some countries. Nevertheless, the marketing of biosimilar medicines has more than 10 years of experience in the EU, and, during this time, biosimilars have maintained safety, efficacy, and quality throughout the process.
In reality, the competent authority for the authorisation of biosimilar medicinal products is the same as for biological reference medicinal products and mandates rigour and compliance with the required criteria for safety, efficacy, and quality. Regarding pharmacovigilance, biosimilar medicines, as well as reference biological medicines, are identified as a group of medicines that require additional monitoring. A risk management plan presents the identified risks, minimisation measures, and strategies to collect immunogenicity data.
A much-discussed aspect among scientists refers to interchangeability and substitution, which must be analysed case by case; it is the obligation of health professionals to be informed about the benefits and risks that such a change may bring to the patient. To this end, the study of the patient and the clinical analysis are essential for adequate and effective medical advice in order to consider the best therapeutic option for the patient in question, according to the stage of the disease.
Yet another challenge in this area is to prove that the extrapolation of data is preserved, which is easily corroborated with all the justification submitted to the EMA and additional clinical trials requested, as well as the inclusion of additional safety measures for monitoring these medicines to ensure their safety.
Understanding the implications of biosimilars and their dissemination is crucial for investment decisions. Deloitte has conducted a study on market entry strategies for biosimilar medicines to explore the investment opportunities available to pharmaceutical companies in this field. Regarding the goals and objectives defined, one should reflect on the real value of the business, keeping in mind the global vision of biosimilars, including the financial and non-financial goals. Next, it is crucial to understand which countries and regions can be considered and the potentials for this market segment and also substantiate which are the most attractive therapeutic areas, specifying what the missing needs in this area are. Bearing in mind the possibility of the existence or emergence of competing biosimilars and new commercial partners, it is important to reflect on their influence on the competitive market and, consequently, the prices charged, which would influence the cost and profit ratio.
6.2. Role of Biosimilar Medicines
Biosimilar medicines today are a reality. They represent a complex and innovative world but with great potential to acquire a promising and prominent role in society. It is expected that investment will be made in the improvement of legislation regarding biosimilars so that the desirable and necessary harmonisation of guidelines becomes a practical and not just a theoretical reality.
Hitherto, biosimilar medicines have demonstrated their importance and efficacy in the fight against diseases and mainly for the well-being of individuals who for various reasons have developed a disease. This contribution is poised to significantly enhance the quality of life for people worldwide. As patents on biological medicines expire, the potential for biosimilar medicines to become a more equitable and sustainable state-of-the-art treatment will become increasingly evident and achievable.
References
- European Medicines Agency. Biosimilar Medicines—An Informative Guide for Health Professionals; European Medicines Agency: Amsterdam, The Netherlands, 2019.
- Longstaff, C.; Whitton, C.M.; Stebbings, R.; Gray, E. How do we assure the quality of biological medicines? Drug Discov. Today 2009, 14, 50–55.
- Portela, E.M.D.C.C.; Sinogas, C.; Almeida, F.A.D.; Baptista-Leite, R.; Castro-Caldas, A. Biologicals and Biosimilars: Gaps in the Pharmacovigilance System in Portugal. Acta Med. Port. 2017, 30, 205–212.
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues; European Medicines Agency: Amsterdam, The Netherlands, 2014.
- Prinja, S.; Pandav, C.S. Economics of COVID-19: Challenges and the way forward for health policy during and after the pandemic. Indian J. Public Health 2020, 64, 231–233.
- European Medicines Agency. O Sistema Regulador Europeu de Medicamentos—Uma Abordagem Coerente à Regulação de Medicamentos na União Europeia; European Medicines Agency: Amsterdam, The Netherlands, 2016.
- Gonçalves, J.; Araújo, F.; Cutolo, M.; Fonseca, J.E. Biosimilar monoclonal antibodies: Preclinical and clinical development aspects. Clin. Exp. Rheumatol. 2016, 34, 698–705.
- Kirchhoff, C.F.; Wang, X.Z.M.; Conlon, H.D.; Anderson, S.; Ryan, A.M.; Bose, A. Biosimilars: Key regulatory considerations and similarity assessment tools. Biotechnol. Bioeng. 2017, 114, 2696–2705.
- Allocati, E.; Godman, B.; Gobbi, M.; Garattini, S.; Banzi, R. Switching Among Biosimilars: A Review of Clinical Evidence. Front. Pharmacol. 2022, 13, 917814.
- World Health Organization. Guidelines on Evaluation of Biosimilars; Replacement of Annex 2 of WHO Technical Report Series, No. 977; WHO: Geneva, Switzerland, 2022.
- Blackstone, E.A.; Joseph, P.F. The Economics of Biosimilars. Am. Health Drug Benefits 2013, 6, 469–478.
- Gonçalves, J. Medicamentos Biossimilares: O Estado da Arte. Lidel—Edico̧ẽs Técnicas, Lda. 2018. Available online: https://books.google.pt/books?id=YZ3lvgEACAAJ (accessed on 17 January 2024).
- FDA. Biosimilar Development Process. Available online: https://www.fda.gov/files/drugs/published/Biosimilar-Development-Process.pdf (accessed on 17 January 2024).
- Desanvicente-Celis, Z.; Gomez-Lopez, A.; Anaya, J.M. Similar biotherapeutic products: Overview and reflections. Immunotherapy 2012, 4, 1841–1857.
- Dranitsaris, G.; Amir, E.; Dorward, K. Biosimilars of biological drug therapies. Drugs 2011, 71, 1527–1536.
- FDA US. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product; FDA: Silver Spring, MD, USA, 2015.
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. mAbs 2010, 2, 480–499.
- Kozlowski, S. US FDA Perspectives on Biosimilar Biological Products; IBBR University of Maryland: Rockville, MD, USA, 2014.
- Genazzani, A.A.; Biggio, G.; Caputi, A.P.; Del Tacca, M.; Drago, F.; Fantozzi, R.; Canonico, P.L. Biosimilar drugs: Concerns and opportunities. BioDrugs 2007, 21, 351–356.
- Ahmad, A.S.; Olech, E.; McClellan, J.E.; Kirchhoff, C.F. Development of Biosimilars. Semin. Arthritis Rheum. 2016, 45, S11–S18.
- Amgen Biosimilars. Developing Biosimilars: The Process and Quality Standards. 2017. Available online: https://www.amgenoncology.com/resources/developing_biosimilars-USA-BIO-047538.pdf (accessed on 17 January 2024).
- Mellstedt, H.; Niederwieser, D.; Ludwig, H. The challenge of biosimilars. Ann. Oncol. 2008, 19, 411–419.
- Roger, S.D. Biosimilars: How similar or dissimilar are they? Nephrology 2006, 11, 341–346.
- Vulto, A.G.; Jaquez, O.A. The process defines the product: What really matters in biosimilar design and production? Rheumatology 2017, 56, iv14–iv29.
- Lowe, D.; Dudgeon, K.; Rouet, R.; Schofield, P.; Jermutus, L.; Christ, D. Aggregation, stability, and formulation of human antibody therapeutics. Adv. Protein Chem. Struct. Biol. 2011, 84, 41–61.
- Warne, N.W. Development of high concentration protein biopharmaceuticals: The use of platform approaches in formulation development. Eur. J. Pharm. Biopharm. 2011, 78, 208–212.
- Kurki, P.; van Aerts, L.; Wolff-Holz, E.; Giezen, T.; Skibeli, V.; Weise, M. Interchangeability of Biosimilars: A European Perspective. BioDrugs 2017, 31, 83–91.
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues; European Medicines Agency: Amsterdam, The Netherlands, 2014.
- Aapro, M.; Cornes, P.; Sun, D.; Abraham, I. Comparative cost efficiency across the European G5 countries of originators and a biosimilar erythropoiesis-stimulating agent to manage chemotherapy-induced anemia in patients with cancer. Ther. Adv. Med. Oncol. 2012, 4, 95–105.
- Praditpornsilpa, K.; Tiranathanagul, K.; Kupatawintu, P.; Jootar, S.; Intragumtornchai, T.; Tungsanga, K.; Teerapornlertratt, T.; Lumlertkul, D.; Townamchai, N.; Susantitaphong, P.; et al. Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011, 80, 88–92.
- European Medicines Agency. Guideline on Immunogenicity Assessment of Therapeutic Proteins; European Medicines Agency: Amsterdam, The Netherlands, 2017.
- Weise, M.; Bielsky, M.C.; De Smet, K.; Ehmann, F.; Ekman, N.; Giezen, T.J.; Gravanis, I.; Heim, H.-K.; Heinonen, E.; Ho, K.; et al. Biosimilars: What clinicians should know. Blood 2012, 120, 5111–5117.
- Perpoil, A.; Grimandi, G.; Birklé, S.; Simonet, J.-F.; Chiffoleau, A.; Bocquet, F. Public Health Impact of Using Biosimilars, Is Automated Follow up Relevant? Int. J. Environ. Res. Public Health 2021, 18, 186.
- Vaz, A.F.; Alcobia, A.; Ribeiro, C.F.; Pinto, C.G.; Canhão, H.; Cruz, J.P.; da Silva, J.A.; Matos, L.; Amaral, M.; Miranda, N.; et al. Medicamentos biossimilares. Rev. Port. Farmacoter. 2013, 5, 44–51.
- Sharma, A.; Khante, S.; Mahadik, K.R.; Gaikwad, V.L. Regulatory Perspective of International Agencies for Development of Biosimilar Products (Monoclonal Antibodies): An Overview. Ther. Innov. Regul. Sci. 2020, 54, 965–977.
- Guideline, Ich Harmonised Tripartite. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process Q5E. 2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnologicalbiological-products-step-5_en.pdf (accessed on 17 January 2024).
- Blank, T.; Netzer, T.; Hildebrandt, W.; Vogt-Eisele, A.; Kaszkin-Bettag, M. Safety and toxicity of biosimilars—EU versus US regulation. GaBI J. 2013, 2, 144–150.
- Schellekens, H.; Moors, E. Clinical comparability and European biosimilar regulations. Nat. Biotechnol. 2010, 28, 28–31.
- Lee, J.F.; Litten, J.B.; Grampp, G. Comparability and biosimilarity: Considerations for the healthcare provider. Curr. Med. Res. Opin. 2012, 28, 1053–1058.
- Mccamish, M.; Woollett, G. The continuum of comparability extends to biosimilarity: How much is enough and what clinical data are necessary? Clin. Pharmacol. Ther. 2013, 93, 315–317.
- Mascarenhas-Melo, F.; Diaz, M.; Gonçalves, M.B.S.; Vieira, P.; Bell, V.; Viana, S.; Nunes, S.; Paiva-Santos, A.C.; Veiga, F. An Overview of Biosimilars—Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals 2024, 17, 235.
- Fernandes, J.P.P.; Gonçalves, J.P. Segurança e Eficácia Clínica na Substituição de Medicamentos Biossimilares. Rev. Port. Farmacoter. 2015, 7, 7–27.
- European Generics Medicines Association. Biosimilars Handbook. Available online: https://www.medicinesforeurope.com/wp-content/uploads/2016/03/EGA_BIOSIMILARS_handbook_en.pdf (accessed on 17 January 2024).
- Braun, J.; Kudrin, A. Switching to biosimilar infliximab (CT-P13): Evidence of clinical safety, effectiveness and impact on public health. Biologicals 2016, 44, 257–266.
- Oliveira, R.; Aires, T. Biossimilares: Velhas Questões, Novos Desafios. Gaz. Méd. 2018, 3, 106–111.
- Market Review. European Biosimilar Medicine Markets 2020; Medicines for Europe: Brussels, Belgium, 2020; Available online: https://www.medicinesforeurope.com/wpcontent/uploads/2021/03/Biosimilar%20Market%20Review-Final.pdf (accessed on 17 January 2024).
- European Medicines Agency. Statement on the Scientific Rationale Supporting Interchangeability of Biosimilar Medicines in the EU. 2022. Available online: https://www.ema.europa.eu/en/news/biosimilar-medicines-can-be-interchanged (accessed on 17 January 2024).
- Tsiftsoglou, A.S.; Ruiz, S.; Schneider, C.K. Development and Regulation of Biosimilars: Current Status and Future Challenges. BioDrugs 2013, 27, 203–211.
- Daller, J.R. Biosimilars: A consideration of the regulations in the United States and European union. Regul. Toxicol. Pharmacol. RTP 2016, 76, 199–208.
- European Medicines Agency. Guideline on Similar Biological Medicinal Products; European Medicines Agency: Amsterdam, The Netherlands, 2014.
- Committee for Medicinal Products for Human Use. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues; European Medicines Agency: London, UK, 2006.
- Kurki, P.; Ekman, N. Biosimilar regulation in the EU. Expert Rev. Clin. Pharmacol. 2015, 8, 649–659.
- Gherghescu, I.; Delgado-Charro, M.B. The biosimilar landscape: An overview of regulatory approvals by the EMA and FDA. Pharmaceutics 2020, 13, 48.
- Jordan, J.B.; Christl, L. FDA Biosimilar Action Plan: Could improving pharmacovigilance of biologics improve patient and physician confidence in biosimilars? Expert Opin. Drug Saf. 2020, 19, 229–232.
- Food and Drug Administration. Purple Book. Database of Licensed Biological Products. Available online: https://purplebooksearch.fda.gov/ (accessed on 17 January 2024).
- Christl, L.A.; Woodcock, J.; Kozlowski, S. Biosimilars: The US regulatory framework. Annu. Rev. Med. 2017, 68, 243–254.
- Yamaguchi, T.; Arato, T. Quality, safety and efficacy of follow-on biologics in Japan. Biologicals 2011, 39, 328–332.
- Generics. Initiative, Biosimilars. Biosimilars Approved in Japan. Posted 07/03/2014. 2015. Available online: http://gabionline.net/Biosimilars/General/Biosimilars (accessed on 17 January 2024).
- Kalaiselvan, V.; Srivastava, S.; Singh, A.; Gupta, S.K. Pharmacovigilance in India: Present Scenario and Future Challenges. Drug Saf. 2019, 42, 339–346.
- Arato, T. Japanese regulation of biosimilar products: Past experience and current challenges. Br. J. Clin. Pharmacol. 2016, 82, 30–40.
- Meher, B.R.; Balan, S.; Mohanty, R.R.; Jena, M.; Das, S. Biosimilars in India; current status and future perspectives. J. Pharm. Bioallied Sci. 2019, 11, 12.
- Health Canada. Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs); Health Canada: Ottawa, ON, Canada, 2010.
- Declerck, P.; Danesi, R.; Petersel, D.; Jacobs, I. The language of biosimilars: Clarification, definitions, and regulatory aspects. Drugs 2017, 77, 671–677.
- Azevedo, V.F.; Sandorff, E.; Siemak, B.; Halbert, R.J. Potential regulatory and commercial environment for biosimilars in Latin America. Value Health Reg. Issues 2012, 1, 228–234.
- De Assis, M.R.; Pinto, V. Strengths and weaknesses of the Brazilian regulation on biosimilars: A critical view of the regulatory requirements for biosimilars in Brazil. Ther. Adv. Musculoskelet. Dis. 2018, 10, 253–259.
- González-Ramírez, R.; Castañeda-Hernández, G. The challenges of developing and commercializing biosimilars in Latin America. Pharm. Pat. Anal. 2019, 8, 221–224.
- Association of Southeast Asian Nations. Bureau of East Asian and Pacific Affairs. U.S. Department of State. 2019. Available online: https://www.state.gov/association-of-southeast-asian-nations-asean/ (accessed on 17 January 2024).
- Fortune Business Insights. Market Rearch Report. Biosimilars Market Size. 2024. Available online: https://www.fortunebusinessinsights.com/biosimilars-market-108928 (accessed on 17 January 2024).
- Remicade European Medicines. 2022. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/Remicade#:~:text=Remicade%20is%20an%20anti%E2%80%91inflammatory%20medicine.%20It%20is%20usually,%28a%20medicine%20that%20acts%20on%20the%20immune%20system%29%3B (accessed on 17 January 2024).
- Remicade. Infliximab. Jassen Biotecnh. Available online: https://www.Remicade.com/ (accessed on 17 January 2024).
- Vieira, I.; Sousa, J.J.; Vitorino, C. Paediatric Medicines—Regulatory Drivers, Restraints, Opportunities and Challenges. J. Pharm. Sci. 2021, 110, 1545–1556.
- Needs for Paediatric Medicines. EMA: European Medicines Agency. 2020. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/paediatric-medicines/needs-paediatric-medicines (accessed on 17 January 2024).
- Narula, N.; Wong, E.C.; Dulai, P.S.; Sengupta, N.K.; Marshall, J.K.; Colombel, J.F.; Reinisch, W. Comparative efficacy and rapidity of action for infliximab vs ustekinumab in biologic naive Crohn’s disease. Clin. Gastroenterol. Hepatol. 2022, 20, 1579–1587.e2.
- Baldassano, R.; Braegger, C.P.; Escher, J.C.; DeWoody, K.; Hendricks, D.F.; Keenan, G.F.; Winter, H.S. Infliximab (REMICADE) therapy in the treatment of pediatric Crohn’s disease. Am. J. Gastroenterol. 2003, 98, 833–838.
- Hermosilla, J.; Sánchez-Martín, R.; Pérez-Robles, R.; Salmerón-García, A.; Casares, S.; Cabeza, J.; Cuadros-Rodríguez, L.; Navas, N. Comparative stability studies of different infliximab and biosimilar CT-P13 clinical solutions by combined use of physicochemical analytical techniques and enzyme-linked immunosorbent assay (ELISA). BioDrugs 2019, 33, 193–205.
- Remicade. EPAR—Procedural Steps Taken before Authorisation; European Medicines Agency: Amsterdam, The Netherlands, 2005.
- Inflectra. EPAR—Public Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2013.
- Remsima. EPAR—Public Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2013.
- Flixabi. EPAR—Public Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2016.
- Zessly. EPAR—Public Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2018.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
05 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No