Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Carlos Sinogas and Version 2 by Camila Xu.
By definition, biosimilar medicinal products are biological medicinal products that are similar to other biological medicinal products that are already on the market—the reference medicinal products.
- biosimilar medicines
- regulatory perspective
- European Medicines Agency
1. Introduction
Biological medicinal products are defined as active substances obtained from a biological source, such as living cells or organisms, most of which are composed of proteins. Manufacturing these products involves complex biotechnological processes, setting them apart from those obtained through chemical means. Due to their intricate molecular structure and inherent variability, biological medicinal products undergo stringent quality and safety assessments [1][2][1,2]. These medicines typically contain one or more active substances sourced from biological materials. Reference biological medicines undergo a thorough evaluation based on comprehensive technical and scientific documentation covering aspects of quality, safety, and efficacy [3].
2. Definition and Characterisation of Biosimilars
According to the EMA, a biosimilar medicine is considered to be a medicine highly similar to another biological medicine that is already marketed in the EU—a reference medicine. Since biosimilars are a type of biological medicinal product, all relevant characteristics of biological medicinal products apply to them. Biosimilar medicinal products are molecules of high molecular weight with high complexity and are produced in cells or transgenic organisms [1]. A biosimilar medicine is developed to be very similar to its reference medicine in terms of quality, safety, and efficacy. Although there may be minor differences due to the complex nature and production methods, the active ingredients of a biosimilar medicine and its biological reference medicine are essentially the same biological substance [4]. In terms of similarity, the biosimilar medicinal product has physical, chemical, and biological properties highly similar to the reference medicine, particularly in terms of safety and efficacy. As for the existing variability, this may exist on a small scale, and it is crucial to demonstrate that it does not affect the safety and efficacy of a biosimilar. As for general standards, all biosimilar medicinal products comply with the same quality, safety, and efficacy standards as any other medicinal product [1]. However, being biological products, there is an associated variability that prevents the biosimilar medicine from being exactly equal to the reference medicine [5][6][5,6]. For the safety and efficacy profile to be maintained, legislation mandates the studies to be conducted so that a biosimilar medicine can demonstrate similarities in terms of quality, safety, and efficacy to its reference medicine and that there are no significant differences [7]. Table 1 shows the comparison between the data requirements for the approval of a biosimilar and a reference medicinal product [1].Table 1. Data requirements for the approval of a biosimilar and a reference medicinal product. (Adapted from [1]).
| Reference Medicinal Product | Biosimilar Medicinal Product |
|---|---|
| Risk Management Plan | Risk Management Plan |
Clinical studies:
|
Comparative clinical studies:
|
| Non-clinical studies | Comparative non-clinical studies |
| Pharmaceutical quality studies | Comparative quality studies: Pharmaceutical quality studies, namely, full data requirements regarding pharmaceutical quality, as well as additional quality studies comparing the structure and biological activity of the biosimilar medicine with the reference medicine. |
3. Development of Biosimilar Medicines
In terms of development, biosimilars entail costs approximately one hundred times higher than those associated with generic drugs, with a development timeline ranging between seven and eight years, as opposed to the two to four years typically required for generic drugs [11]. The basis of the development of a biosimilar drug is thus an extensive structural and functional characterisation as well as the comparison of the biosimilar drug with the reference drug. The development of a biosimilar medicine starts with defining the fingerprint of the reference medicine and its attributes, establishing the boundaries of potential variability for the biosimilar. Since the manufacturing process of the reference molecule remains undisclosed, a novel process must be devised to ensure alignment with the fingerprint. Such a process requires that the cell culture and purification process conditions are continuously adjusted, exploring new cell lines throughout the development until the highest similarity is achieved. Progress towards clinical trials for the potential biosimilar hinges on achieving full characterization of the molecule, well-defined processes, and confirmed molecular similarity [7][12][13][7,12,13]. The development process for biosimilars is complex, encompassing various stages such as cell line selection, cultivation, production, isolation, purification, and, ultimately, formulation, filling, and finishing. The first decision in the development of a biosimilar is about the cell line creation (Figure 1), being such an important decision for the glycosylation patterns and the determination of the final profile of the biosimilar so that the protein of interest is expressed. The relevant gene is then cloned into a complementary DNA vector, which could be derived from human or microbial cell lines. Generally, the industry opts for Chinese hamster ovary cells for expression cells due to their consistent folding, ability to thrive in suspension, high yield, and stability against changes in pH and oxygen levels [14][15][16][17][14,15,16,17]. Following this step is clone selection, aiming to identify clones that closely mirror the desired product fingerprint. It is crucial to note that no two biologics are identical as each product is manufactured using a unique cell line from the producer [18]. It is important to mention that there are no two identical biologics because each product is produced using an exclusive cell line from the manufacturer [19][20][19,20]. Moving towards the second step in the development of a biosimilar, cultivation and production (Figure 2), the cell line that will produce the protein and originate the biosimilar medicine is expanded in a fermentation medium to obtain a batch of master cells, and these cells are then cultivated and grown in large-scale bioreactors [14][15][22][23][14,15,22,23]. First, thawing of the cells occurs, followed by inoculation under agitation to increase cell density. To ensure cell viability, the cells are maintained in a growth medium fortified with nutrients and supplements [24]. Finally, it is also important to mention that throughout this process, parameters such as the amount of oxygen, lactate production, temperature, pH, and osmolarity are controlled [20][25][20,25].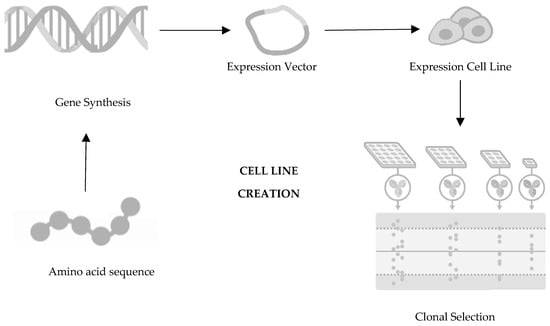
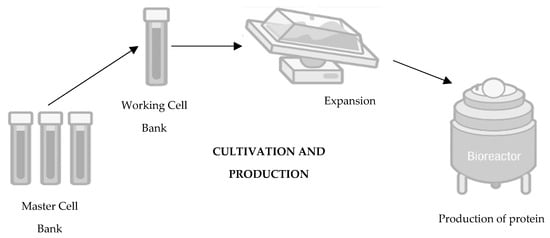
Figure 2.
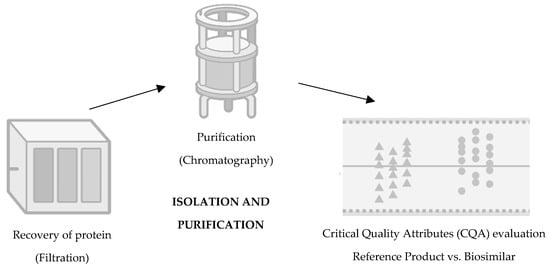
Figure 4.
3.1. Immunogenicity
Concerning immunogenicity, proteins inherently possess the capacity to elicit an undesired immune response. In rare instances, this reaction can lead to severe adverse effects or even diminish efficacy, although such occurrences are infrequent. Immunogenicity is influenced by various factors, including the drug characteristics, external factors associated with treatment, and individual patient- or disease-related factors. Quality issues, such as alterations in production methods, formulations, or packaging, can impact immunogenicity by affecting the drug properties and impurity profile. However, it is crucial to ensure that these changes do not compromise the medicine’s efficacy and safety [29][30][29,30]. It is very unlikely for an adverse immune reaction to occur following the aforementioned modifications as comparability studies indicate no significant increase in impurities or aggregates despite these alterations. Regulatory agencies actively monitor the immunogenicity of biosimilar drugs, employing comparability studies across different batches and utilizing physicochemical and structural analyses alongside functional in vitro assays [31][32][31,32]. The immunogenicity data required for the approval of a biological medicinal product for approval purposes, as highlighted by the EMA, include the incidence, titration, and persistence of antibodies against the biological medicinal product; neutralisation tests; clinical impact assessment; and measures to manage the risk of immunogenic potential. Nevertheless, these data always depend on the type of biological medicine and its intended use and the characteristics of the product itself, among others [1][33][1,33].3.2. Extrapolation
Extrapolation represents a well-established scientific concept, aiming to evaluate the extent to which a biosimilar closely resembles a reference medicinal product in terms of safety and efficacy within a particular therapeutic indication. This similarity allows for the extension of these findings to other approved indications for the reference product. In practical terms, this means that, in some situations, few clinical studies with biosimilars may be carried out since this extrapolation is always supported by scientific evidence from comparability studies. Extrapolation criteria typically include considerations such as the mechanism of action, the target study population, various clinical settings, safety profiles, and immunogenicity data [1][34][1,34]. Concerning the mechanism of action of the active substance, it should be mediated by the same receptor, and additional research may be necessary to demonstrate similarity in behaviour. Depending on the clinical context, variations in dosage, pharmacokinetics, or mode of action may arise, necessitating further studies as the data for one indication may not directly apply to another. Establishing a comparable safety profile for a specific indication is crucial at this stage before extrapolating safety data. Immunogenicity data, being a specific criterion, require thorough substantiation and add complexity to the process [1][35][1,35].3.3. Safety
In addressing the safety of biosimilar medicinal products, a comprehensive approach is essential, from a sound regulatory framework, a risk management plan always in place, post-authorisation safety studies, and continuous safety monitoring. Given that many adverse drug reactions associated with biosimilars can be anticipated based on their pharmacological effects, efforts are made to collect reports of adverse reactions spontaneously. This includes the submission of periodic safety reports and the implementation of additional monitoring mechanisms to detect potential long-term adverse events or with long latency periods [1]. The EU has a consolidated system of monitoring, reporting, assessing, and preventing adverse drug reactions, so the authorities continuously assess the risk–benefit ratio of all medicines and take the necessary measures. When an undertaking applies for the marketing authorisation, it will submit a risk management plan including a pharmacovigilance plan as well as any risk minimisation measures to identify, characterise, and minimise risks. When talking about a risk management plan (RMP) for a biosimilar medicine, it will always be based on the knowledge and experience already gained with the reference medicine. For post-authorisation studies, it has been made possible to monitor known risks and detect adverse drug reactions that only arise after a large number of patients receive treatment over a substantial time period. In addition, it is noteworthy that toxicity studies are used to carry out this safety assessment of biological medicinal products. The main mechanisms of adverse effects of biological drugs are related to exaggerated pharmacology. This phenomenon, known as on-target toxicity, entails the occurrence of adverse pharmacological effects specifically at the intended target site. However, if comparable pharmacological activity has been established in vitro, there is no need to confirm these mechanistic properties in vivo [36]. Examples of unexpected toxicity are scarce. Often, functional differences between these antibodies have a structural basis rooted in variations in the amino acid sequence. These differences are not integrated into the development of similar biological medicines because the amino acid sequence should be the same as that of the reference medicine [37]. Thus, although unexpected toxicity can be found in pre-clinical animal studies during the development of new biological medicinal products, there is no evidence to suggest that such occurrences are associated with biosimilars [12].3.4. Comparability
The fundamental principle of developing a biosimilar is based on the comparability exercise that is carried out between the reference biological medicine and the biosimilar medicine. For such a comparability exercise to be undertaken, comprehensive analyses of the proposed biosimilar and the reference product are required, using sensitive methods. The main objective is to demonstrate that the biosimilar and the reference product are similar at a finished medicinal product level [28]. In this context, several important pillars arise when comparing the reference medicine and the biosimilar medicine: target/quality by clinical trial design, quality (data set for stand-alone) biological and physicochemical comparability, comparative pre-clinical trials, comparative clinical trials, and risk management plan [11]. The comparability exercise is carried out in several steps, namely, quality comparability, non-clinical comparability, and clinical comparability. In the first step, quality comparability, a full characterisation approach needs to be undertaken to compare the physicochemical and biological quality attributes, for instance, the purity of the potential biosimilar medicine. This is performed using a wide range of different state-of-the-art analytical tests, as no single method can characterise all aspects of a product. The development process is also to be modified if there are significant differences found in the analyses until the product generated has a profile that matches the reference product profile. Iterative adjustments are made at every phase of the development process to ensure that the ultimate biosimilar medicine meets the quality standards set by the EMA, aligning with all criteria stipulated for documentation submission, assessment, and marketing authorization. In the second step, non-clinical comparability, non-clinical studies, sometimes called pre-clinical studies, need to be conducted for biosimilar medicines before any clinical trial begins. Generally, non-clinical data for a biosimilar are generated through an abbreviated testing programme or in vivo animal studies, as required by the EMA’s guidelines. On the other hand, non-clinical studies usually include repeated dose toxicity studies as well as pharmacokinetic and pharmacodynamic (PK/PD) studies along with local tolerance testing. For this type of study, similarity criteria should be pre-defined and scientifically justified in order to allow comparability of the support with the reference and detect potential differences between them. In the third step, clinical comparability, clinical tests are also considered comparative in the case of the development of a biosimilar medicinal product. However, such clinical tests are not required to the same extent as would be necessary for a new active substance, taking into account the clinical experience gained from the use of the reference medicinal product. Therefore, it is crucial to consider the nature and characteristics of the medicinal product as well as the therapeutic indications. Another key aspect is to understand how comparable the profile of the biosimilar is to that of the reference product. The closer the biosimilar and reference profiles are and the greater the similarity demonstrated (through appropriate studies such as comparative quality, biological and receptor binding activity analyses, and animal testing), the more easily the clinical trial programme will be accepted by the regulatory authorities. In summary, the clinical comparability exercise starts with pharmacokinetic and/or pharmacodynamic studies, and comparative clinical efficacy and safety trials may still follow. An important step is to document the side effects and to take into consideration the evaluation of immunogenicity for which comparable profiles for the biosimilar and the reference are also required for the clinical safety data assessment [38][39][38,39]. Apart from comparing the quality attributes of both medications, establishing the similarity in biological activity and safety of the biosimilar involves utilizing relevant and sensitive in vitro assays. These assays help identify significant differences in allosteric mechanisms between the two medications across all approved indications [40]. Table 2 outlines the comparability critical attributes and the way the similarity of the products is demonstrated [12][40][12,40].| Comparability Critical Attributes | Similarity Demonstration |
|---|---|
| Protein structure and production quality | Extensive laboratory analysis for all molecular characteristics (multiple lots) |
| Pharmacokinetics, Pharmacodynamics, and Animal Toxicity | In vitro and in vivo assays (carried out on relevant animal species, if further confirmation scans are required for laboratory studies) |
| Pharmacokinetics, Pharmacodynamics, and Animal Toxicity | Phase I clinical studies |
| Clinical Efficacy and Safety | Phase III clinical studies |
| Safety in clinical practice | Risk management plan Phase IV studies Pharmacovigilance |
3.5. Interchangeability
The term interchangeability emerged in the context of the possibility of exchanging a reference biological medicine for its biosimilar, or a biosimilar for another biosimilar, hoping to obtain the same therapeutic effect produced by the option initially used [1][42][1,42]. This exchange can occur in two distinct ways: switch or automatic substitution. As for the “switch” option, this occurs when the prescribing physician decides to exchange one medicine for another that has the same therapeutic purpose. Automatic substitution, on the other hand, refers to a practice carried out at the pharmacy level without consulting the prescribing physician, where the dispensing of the prescribed drug is changed by another equivalent and interchangeable drug [1][43][1,43]. These issues have been the subject of some controversy, despite the existence of several studies that prove the safety and effectiveness of this practice [44]. In terms of pharmacovigilance, the traceability of the biological medicine involved in the potential adverse reaction is very important, so the same brand of medicine should be kept for as long as the reaction is expected to occur. The change between biosimilar biological medicines should respect a minimum period of time that safeguards their traceability. This information can be found in the National Medicines Formulary and, when this is not mentioned, that period should not be less than 6 months. Switching between different brands of the same biological medicine must be articulated with the clinical services involved, with respect to the precautionary principle and in accordance with the therapeutic indications for each situation. This position is reviewed whenever applicable scientific evidence becomes available. Therefore, what is the EMA’s position on the issue of switching the biosimilar as a reference medicine? Previously, the EMA positioning was directed to the independent deliberation by each member state. In Portugal, the decision regarding therapeutic switching is made by INFARMED I.P. in conjunction with the National Pharmacy and Therapeutics Commission. They stipulate that the switch should occur only after the specified minimum duration to guarantee medicine traceability. However, automatic substitution is not currently permitted in Portugal [1][45][46][1,45,46]. Note, however, that in September 2022, as reported on the EMA’s website, the EMA and the Heads of Medicines Agencies (HMA) released a joint statement affirming the interchangeability of biosimilar medicines approved in the EU with their reference medicine or an equivalent biosimilar. This unified stance serves to standardize the approach across the EU [47].4. Regulatory Framework for the Development of Biosimilars
The marketing authorisation for biosimilar medicinal products, via the EMA, is obtained through a centralised procedure, in which the Agency evaluates the medicinal products for the purpose of authorising their marketing. Comparative characteristics studies evaluate the composition, physical properties, protein structure, purity, isoforms, or impurities that derive from the product, as well as biological activity. The production process between the reference biological drug and the biosimilar may, however, present differences. Concerning the regulatory framework for the development of biosimilars, the perspective of the approval process via the EMA and FDA as well as other regulatory agencies may differ in certain aspects [6][48][6,48].4.1. European Medicines Agency
All biotechnologically produced medicinal products are approved in the EU by the EMA through a centralised procedure. There are certainly some biosimilars, such as low-molecular-weight heparins obtained from the swine intestinal mucosa, which are approved at the national level; however, mostly, and since they resort to biotechnology for production, they are approved by the centralized procedure. First, the entity submits the marketing authorisation application to the EMA. The data are then evaluated by the scientific committees, the Committee for Medicinal Products for Human Use (CHMP) and the Pharmacovigilance Risk Assessment Committee (PRAC), as well as by experts in biological medicinal products and biosimilar specialists. Subsequently, this analysis gives rise to a scientific opinion sent to the European Commission (EC). If applicable, it is for the EC to grant marketing authorisation in the EU and it is valid in all EU member states [1]. The EMA was the first organisation to delineate a specific regulatory route for biosimilar medicines; this happened in October 2005. The EMA guidelines state that the approval of biosimilars is based on comparability studies, through the characterisation of the protein structure of the biosimilar, as well as its efficacy, safety, and immunogenicity. In addition to these aspects, it is considered essential to carry out in vitro tests, impurity profile analyses, and pharmacokinetic and pharmacodynamic studies, as well as pharmacovigilance monitoring after its approval on the market [49]. There are several important requirements according to the Agency, including the importance of demonstrating robustness and consistency in its manufacturing process, as well as details for additional non-clinical and clinical studies to attest to the comparability. A key point highlighted by the EMA is that the clinical benefit has already been proven by the innovative product; so, the goal of a biosimilar is to demonstrate similarity to the innovative product, in terms of strength, active substance, route of administration, and pharmaceutical form, and not the clinical benefit itself. In order to compile all legislation concerning biological medicines, the EMA has adopted several more individualised guidelines for each type of biosimilar medicine, as presented in Table 3. The general guidelines include the Guideline on similar biological medicinal products, the Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues, and the Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: clinical and non-clinical issues [4][28][31][50][51][4,28,31,50,51]. In addition, the EMA has also developed more specific guidelines: the Guideline on biosimilar medicines containing recombinant granulocyte colony-stimulating factor, Guideline on clinical and non-clinical development of similar medicines containing low-molecular-weight heparins, Guideline on clinical and non-clinical development of similar medicines containing recombinant human insulin and insulin analogue, Guideline on clinical and non-clinical development of similar medicines containing beta interferon, Guideline on clinical and non-clinical development of similar medicines containing monoclonal antibodies: ethical and non-clinical issues, Guideline on clinical and non-clinical development of similar medicines containing recombinant erythropoietin, Guideline on clinical and non-clinical development of similar medicines containing recombinant follicle-stimulating hormone, and Guideline on clinical and non-clinical development of similar medicines containing somatropin [4][28][31][50][51][4,28,31,50,51]. Finally, two other relevant guidelines on the comparability of biotechnology-derived medicinal products after a change in the manufacturing process exist: clinical and non-clinical issues and another one on ICH Q5E biological/biotechnology products subject to changes in their manufacturing processes: comparability of biological/biotechnology products [4][28][31][50][51][4,28,31,50,51].Table 3.
| Type of Guideline | Guideline Title |
| General | Guideline on similar biological medicinal products. |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues. | |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: clinical and non-clinical issues. | |
| Specific | Guideline on biosimilar medicines containing recombinant granulocyte colony-stimulating factor. |
| Guideline on clinical and non-clinical development of similar medicines containing low-molecular-weight heparins. | |
| Guideline on clinical and non-clinical development of similar medicines containing recombinant human insulin and insulin analogue. | |
| Guideline on clinical and non-clinical development of similar medicines containing beta interferon. | |
| Guideline on clinical and non-clinical development of similar medicines containing monoclonal antibodies: ethical and non-clinical issues. | |
| Guideline on clinical and non-clinical development of similar medicines containing recombinant erythropoietin. | |
| Guideline on clinical and non-clinical development of similar medicines containing the recombinant follicle-stimulating hormone. | |
| Guideline on clinical and non-clinical development of similar medicines containing somatropin. | |
| Others | Comparability of biotechnology-derived medicinal products after a change in manufacturing process: clinical and non-clinical issues. |
| ICH Q5E biological/biotechnology products subject to changes in their manufacturing processes: comparability of biological/biotechnology products. |
4.2. Food and Drug Administration
In the United States, the creation of guidelines for biosimilars did not happen as early as in the EU. The FDA has only recently started formulating distinct guidelines for the approval of biosimilar drugs. At the level of important factors to consider at the time of the FDA’s approval process, the need for robustness in the manufacturing process, the similarity of the protein structure, the extent of understanding of the mechanism of action, appropriate pharmacodynamics trials, comparative pharmacokinetic data, immunogenicity data, and clinical data regarding the innovative product are highlighted [49]. In the initial phase, the biosimilar medicinal product entity presents a series of data comparing the candidate medicinal product with the reference medicinal product in order to demonstrate similarity. First, the data are evaluated, starting with a detailed analytical characterisation, at the structural and functional level, as well as a comparison, moving to animal studies, and, if necessary, comparative clinical studies [18]. The submission of a biosimilar medicinal product will include some data to demonstrate such similarity with the reference medicinal product, from analytical studies demonstrating that the biosimilar medicinal product is highly similar to the reference medicinal product to toxicity assessment studies. Another aspect refers to a clinical study to demonstrate the safety, purity, and potency of the proposed biosimilar product in one or more of the indications for which the reference product is licensed. This typically includes the evaluation of immunogenicity, pharmacokinetics (PK), and, in some cases, pharmacodynamics (PD) and may also include a comparative clinical study [53]. More practically, the application for marketing authorisation is made to the FDA and submitted through section 351(k) of the Public Health Service Act (PHS). This section allows the biosimilar medicinal product to be approved for all therapeutic indications of the reference medicinal product. This happens several times when discussing the speed of the drug approval process in the United States. This situation is due to the fact that, in 2010, a change was made to the Biologics Price Competition and Innovation Act, enabling the existence of a shortened route for the regulation of biosimilar drugs in the USA. However, it is important to point out that such a change does not diminish the required standards of safety, purity, and efficacy. As with the EMA, the FDA requires a rigid, sequential approach with rigorous comparability tests, from physical-chemical, analytical, functional, and non-clinical and clinical assessments. In 2014, the Purple Book was published by the FDA, where reference drugs and corresponding biosimilars are found, presenting the medicines authorised for commercialisation in the USA [54][55][54,55]. In 2015, the FDA published three guidelines, granting industry guidelines for the development of biosimilar drugs. These include scientific assessments in the demonstration of similarity, considerations about the quality for the demonstration of similarity with the reference medicinal product, and a set of questions and answers on the evaluation of the drugs in question [16]. In 2018, the FDA’s Biosimilar Action Plan (BAP) was published, providing information regarding actions to stimulate the development of biosimilars. Briefly, the BAP analyses the phases so that development and approval are more effective, clarifying regulation and developing support materials for healthcare professionals and patients to demystify the issue and increase confidence. Nevertheless, the BAP does not address issues related to improvements in pharmacovigilance [54][56][54,56]. Table 4 summarises the key criteria for developing a biosimilar medicine through FDA legislation. The FDA’s determination of similarity is based on the totality of evidence provided in the marketing application for FDA review. The data set in the marketing application includes extensive analytical comparison to show that the proposed biosimilar and the reference products are extremely similar in structure and function. Animal, human pharmacological, immunological, and other data are added as needed to the analytical data in a stepwise manner to provide the necessary information with the ultimate goal of demonstrating similarity [13].Table 4.
| FDA advice on the scope and extent of testing during development | Highly Similar | The totality of the evidence | |
| Analytical Studies | Quality characteristics assessment, using state-of-the-art technologies and multiple different tests for the same characteristic, to determine whether the proposed biosimilar is highly similar to the reference product. Identification of differences in quality characteristics, if applicable, between the reference product and the proposed biosimilar (examples of quality characteristics may include structure and bioactivity). Evaluation of the potential impact of any observed differences. |
||
| Toxicity’s Assessment | |||
| Animal Studies | Support safety decision making before human exposure to the proposed biosimilar. Additional support to demonstrate similarity (but not always necessary). |
||
| No Clinically Meaningful Differences | |||
| Human PK and PD Studies | Comparison between the pharmacokinetics (exposure) and, if applicable, the pharmacodynamic (response) profiles of the proposed reference and biosimilar product to support a conclusion of similar efficacy and safety. Human PK and PD studies are generally considered to be the most sensitive data element to support a demonstration of no clinically meaningful differences. |
||
| Immunogenicity Assessment |
Comparison between the incidence and severity of immune responses generated with the reference product and the proposed biosimilar. Immunogenicity assessment is generally included as part of all clinical studies. |
||
| Additional Clinical Studies | Additional clinical studies when residual uncertainties remain about demonstrating that there are no clinically meaningful differences after conducting the above-mentioned studies. | ||
| Experience with the Reference Medicine | |||
| Biosimilarity: According to the general criteria defined by the FDA, the demonstration of biosimilarity is based on the totality of evidence provided when submitting a marketing application for FDA review. The presented data include an extensive analytical comparison in order to show that the proposed biosimilar medicine and the respective reference product are extremely similar in terms of structure and function. Moreover, animal, human pharmacological, immunological, and clinical data are enhanced as necessary to the analytical data, in a stepwise approach to provide the required information to demonstrate biosimilarity. | |||
