 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mansi Patel | -- | 5435 | 2024-02-29 06:44:33 | | | |
| 2 | Jason Zhu | Meta information modification | 5435 | 2024-03-01 10:58:39 | | |
Video Upload Options
Developmental biology is intricately regulated by epigenetics and metabolism but the mechanisms are not completely understood. The situation becomes even more complicated during diseases where all three phenomena are dysregulated. A salient example is COVID-19, where the death toll exceeded 6.96 million in 4 years, while the virus continues to mutate into different variants and infect people. Early evidence during the pandemic showed that the host’s immune and inflammatory responses to COVID-19 (like the cytokine storm) impacted the host’s metabolism, causing damage to the host’s organs and overall physiology. The involvement of angiotensin-converting enzyme 2 (ACE2), the pivotal host receptor for the SARS-CoV-2 virus, was identified and linked to epigenetic abnormalities along with other contributing factors.
1. Introduction
2. SARS-CoV-2 Induces Metabolic Reprogramming and Epigenetic Changes
3. COVID-19 Is Associated with Accelerated Epigenetic Aging and Hence Development
4. Epigenetic Regulation of Viral Pathogenicity Suggests Epi-Drugs as a Therapeutic Approach against COVID-19
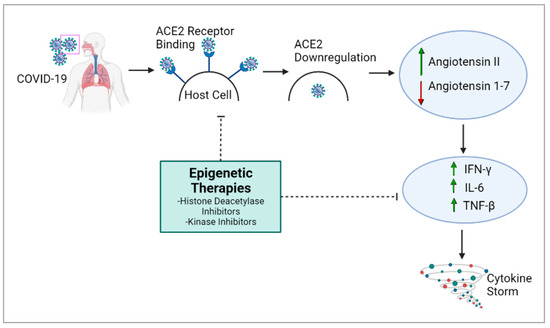
5. Epigenetic Therapies May Help to Mitigate COVID-19 Severity
6. Vitamin D Has a Plausible Protective Effect against COVID-19
7. Overview of Metabolic Abnormalities Associated with COVID-19
8. Abnormal Metabolism and Diabetes Are Often Manifested in COVID-19
9. Hyperglycemia Is Associated with COVID-19 Severity
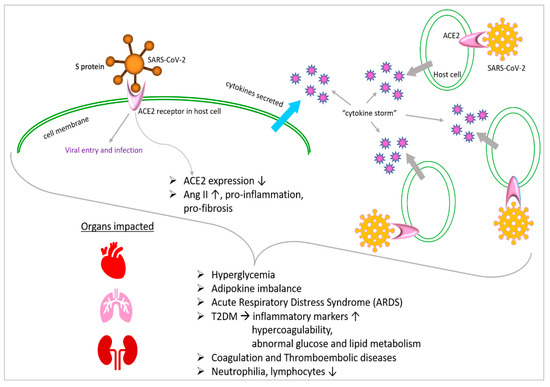
10. COVID-19 Affects Adipokines with Impacts on Glucose Metabolism
11. COVID-19 Affects Metabolism through Interactions with Angiotensin
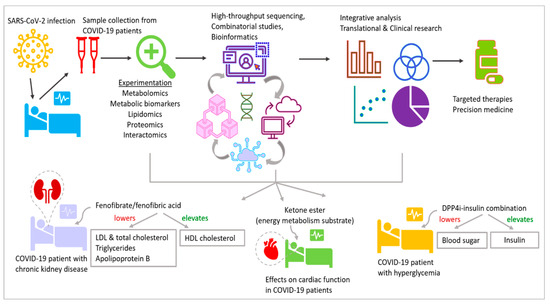
12. Metabolism-Related Therapeutics Could Be Promising against COVID-19
13. Inflammatory Immune Response in Diabetic COVID-19 Patients Is Deleterious
14. Immunometabolic Phenotyping Reveals T Cell and Myeloid Cell Populations Unique to Severe COVID-19
15. Hyperglycemia Associated with COVID-19 Impacts Blood Coagulation
References
- Spinney, L. How pandemics shape social evolution. Nature 2019, 574, 324–326.
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160.
- Prajapati, J.; Rao, P.; Poojara, L.; Goswami, D.; Acharya, D.; Patel, S.K.; Rawal, R.M. Unravelling the antifungal mode of action of curcumin by potential inhibition of CYP51B: A computational study validated in vitro on mucormycosis agent, Rhizopus oryzae. Arch. Biochem. Biophys. 2021, 712, 109048.
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298.
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135.
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther. Nucleic Acids 2019, 17, 24–37.
- Santorelli, L.; Caterino, M.; Costanzo, M. Dynamic Interactomics by Cross-Linking Mass Spectrometry: Mapping the Daily Cell Life in Postgenomic Era. OMICS 2022, 26, 633–649.
- Mar, D.; Babenko, I.M.; Zhang, R.; Noble, W.S.; Denisenko, O.; Vaisar, T.; Bomsztyk, K. A High-Throughput PIXUL-Matrix-Based Toolbox to Profile Frozen and Formalin-Fixed Paraffin-Embedded Tissues Multiomes. Lab. Investig. 2024, 104, 100282.
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032.
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol. 2020, 5, 751–753.
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637.
- Li, S.; Ma, F.; Yokota, T.; Garcia, G., Jr.; Palermo, A.; Wang, Y.; Farrell, C.; Wang, Y.C.; Wu, R.; Zhou, Z.; et al. Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2-induced systemic toxicity. JCI Insight 2021, 6, r145027.
- Sarkar, S.; Sen, R. Insights into Cardiovascular Defects and Cardiac Epigenome in the Context of COVID-19. Epigenomes 2022, 6, 13.
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981.
- Levin, A.T.; Hanage, W.P.; Owusu-Boaitey, N.; Cochran, K.B.; Walsh, S.P.; Meyerowitz-Katz, G. Assessing the age specificity of infection fatality rates for COVID-19: Systematic review, meta-analysis, and public policy implications. Eur. J. Epidemiol. 2020, 35, 1123–1138.
- Gupta, S.; Hayek, S.S.; Wang, W.; Chan, L.; Mathews, K.S.; Melamed, M.L.; Brenner, S.K.; Leonberg-Yoo, A.; Schenck, E.J.; Radbel, J.; et al. Factors Associated With Death in Critically Ill Patients With Coronavirus Disease 2019 in the US. JAMA Intern. Med. 2020, 180, 1436–1447.
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26.
- Yu, M.; Hazelton, W.D.; Luebeck, G.E.; Grady, W.M. Epigenetic Aging: More Than Just a Clock When It Comes to Cancer. Cancer Res. 2020, 80, 367–374.
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314.e9.
- Mollica, V.; Rizzo, A.; Massari, F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol 2020, 16, 2029–2033.
- Beyerstedt, S.; Casaro, E.B.; Rangel, E.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170.
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708.
- Dey, A.; Vaishak, K.; Deka, D.; Radhakrishnan, A.K.; Paul, S.; Shanmugam, P.; Daniel, A.P.; Pathak, S.; Duttaroy, A.K.; Banerjee, A. Epigenetic perspectives associated with COVID-19 infection and related cytokine storm: An updated review. Infection 2023, 51, 1603–1618.
- Kgatle, M.M.; Lawal, I.O.; Mashabela, G.; Boshomane, T.M.G.; Koatale, P.C.; Mahasha, P.W.; Ndlovu, H.; Vorster, M.; Rodrigues, H.G.; Zeevaart, J.R.; et al. COVID-19 Is a Multi-Organ Aggressor: Epigenetic and Clinical Marks. Front. Immunol. 2021, 12, 752380.
- AbdelHamid, S.G.; Refaat, A.A.; Benjamin, A.M.; Elmawardy, L.A.; Elgendy, L.A.; Manolly, M.M.; Elmaksoud, N.A.; Sherif, N.; Hamdy, N.M. Deciphering epigenetic(s) role in modulating susceptibility to and severity of COVID-19 infection and/or outcome: A systematic rapid review. Environ. Sci. Pollut. Res. Int. 2021, 28, 54209–54221.
- Gracia-Ramos, A.E. Is the ACE2 Overexpression a Risk Factor for COVID-19 Infection? Arch. Med. Res. 2020, 51, 345–346.
- Takahashi, Y.; Hayakawa, A.; Sano, R.; Fukuda, H.; Harada, M.; Kubo, R.; Okawa, T.; Kominato, Y. Histone deacetylase inhibitors suppress ACE2 and ABO simultaneously, suggesting a preventive potential against COVID-19. Sci. Rep. 2021, 11, 3379.
- Yasmin, R.; Siraj, S.; Hassan, A.; Khan, A.R.; Abbasi, R.; Ahmad, N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediat. Inflamm. 2015, 2015, 201703.
- Patra, S.K.; Szyf, M. Epigenetic perspectives of COVID-19: Virus infection to disease progression and therapeutic control. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166527.
- Li, Y.; Li, H.; Zhou, L. EZH2-mediated H3K27me3 inhibits ACE2 expression. Biochem. Biophys. Res. Commun. 2020, 526, 947–952.
- Foolchand, A.; Mazaleni, S.; Ghazi, T.; Chuturgoon, A.A. A Review: Highlighting the Links between Epigenetics, COVID-19 Infection, and Vitamin D. Int. J. Mol. Sci. 2022, 23, 12292.
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andreo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822.
- de Abajo, F.J.; Rodriguez-Martin, S.; Lerma, V.; Mejia-Abril, G.; Aguilar, M.; Garcia-Luque, A.; Laredo, L.; Laosa, O.; Centeno-Soto, G.A.; Angeles Galvez, M.; et al. Use of renin-angiotensin-aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: A case-population study. Lancet 2020, 395, 1705–1714.
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507.
- Hughes, D.A.; Norton, R. Vitamin D and respiratory health. Clin. Exp. Immunol. 2009, 158, 20–25.
- Lau, F.H.; Majumder, R.; Torabi, R.; Saeg, F.; Hoffman, R.; Cirillo, J.D.; Greiffenstein, P. Vitamin D insufficiency is prevalent in severe COVID-19. medRxiv 2020.
- Tan, C.W.; Ho, L.P.; Kalimuddin, S.; Cherng, B.P.Z.; Teh, Y.E.; Thien, S.Y.; Wong, H.M.; Tern, P.J.W.; Chandran, M.; Chay, J.W.M.; et al. Cohort study to evaluate the effect of vitamin D, magnesium, and vitamin B(12) in combination on progression to severe outcomes in older patients with coronavirus (COVID-19). Nutrition 2020, 79–80, 111017.
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785.
- Stefan, N. Metabolic disorders, COVID-19 and vaccine-breakthrough infections. Nat. Rev. Endocrinol. 2022, 18, 75–76.
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B. Global pandemics interconnected—Obesity, impaired metabolic health and COVID-19. Nat. Rev. Endocrinol. 2021, 17, 135–149.
- Brosh-Nissimov, T.; Orenbuch-Harroch, E.; Chowers, M.; Elbaz, M.; Nesher, L.; Stein, M.; Maor, Y.; Cohen, R.; Hussein, K.; Weinberger, M.; et al. BNT162b2 vaccine breakthrough: Clinical characteristics of 152 fully vaccinated hospitalized COVID-19 patients in Israel. Clin. Microbiol. Infect. 2021, 27, 1652–1657.
- Maestre-Muniz, M.M.; Arias, A.; Mata-Vazquez, E.; Martin-Toledano, M.; Lopez-Larramona, G.; Ruiz-Chicote, A.M.; Nieto-Sandoval, B.; Lucendo, A.J. Long-Term Outcomes of Patients with Coronavirus Disease 2019 at One Year after Hospital Discharge. J. Clin. Med. 2021, 10, 2945.
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the Mystery Surrounding Post-Acute Sequelae of COVID-19. Front. Immunol. 2021, 12, 686029.
- Costanzo, M.; Caterino, M.; Fedele, R.; Cevenini, A.; Pontillo, M.; Barra, L.; Ruoppolo, M. COVIDomics: The Proteomic and Metabolomic Signatures of COVID-19. Int. J. Mol. Sci. 2022, 23, 2414.
- Holman, N.; Knighton, P.; Kar, P.; O’Keefe, J.; Curley, M.; Weaver, A.; Barron, E.; Bakhai, C.; Khunti, K.; Wareham, N.J.; et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 823–833.
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581.
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95.
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e1063.
- Barron, E.; Bakhai, C.; Kar, P.; Weaver, A.; Bradley, D.; Ismail, H.; Knighton, P.; Holman, N.; Khunti, K.; Sattar, N.; et al. Associations of type 1 and type 2 diabetes with COVID-19-related mortality in England: A whole-population study. Lancet Diabetes Endocrinol. 2020, 8, 813–822.
- Lim, S.; Bae, J.H.; Kwon, H.S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30.
- Seidu, S.; Gillies, C.; Zaccardi, F.; Kunutsor, S.K.; Hartmann-Boyce, J.; Yates, T.; Singh, A.K.; Davies, M.J.; Khunti, K. The impact of obesity on severe disease and mortality in people with SARS-CoV-2: A systematic review and meta-analysis. Endocrinol. Diabetes Metab. 2021, 4, e00176.
- Khunti, K.; Kosiborod, M.; Ray, K.K. Legacy benefits of blood glucose, blood pressure and lipid control in individuals with diabetes and cardiovascular disease: Time to overcome multifactorial therapeutic inertia? Diabetes Obes. Metab. 2018, 20, 1337–1341.
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720.
- Mabrey, F.L.; Morrell, E.D.; Wurfel, M.M. TLRs in COVID-19: How they drive immunopathology and the rationale for modulation. Innate Immun. 2021, 27, 503–513.
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schafer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15.
- Iacobellis, G. COVID-19 and diabetes: Can DPP4 inhibition play a role? Diabetes Res. Clin. Pract. 2020, 162, 108125.
- Kokic Males, V. Letter to the editor in response to the article “COVID-19 and diabetes: Can DPP4 inhibition play a role?”. Diabetes Res. Clin. Pract. 2020, 163, 108163.
- Morin, N. Response to COVID-19 and diabetes: Can DPP4 inhibition play a role?—GLP-1 might play one too. Diabetes Res. Clin. Pract. 2020, 164, 108160.
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3319.
- Blanke, C.D. In response: Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3331.
- Fadini, G.P.; Morieri, M.L.; Longato, E.; Avogaro, A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Investig. 2020, 43, 867–869.
- Sun, Y.; Wang, Q.; Yang, G.; Lin, C.; Zhang, Y.; Yang, P. Weight and prognosis for influenza A(H1N1)pdm09 infection during the pandemic period between 2009 and 2011: A systematic review of observational studies with meta-analysis. Infect. Dis. 2016, 48, 813–822.
- Carter, S.J.; Baranauskas, M.N.; Fly, A.D. Considerations for Obesity, Vitamin D, and Physical Activity Amid the COVID-19 Pandemic. Obesity 2020, 28, 1176–1177.
- Remuzzi, A.; Remuzzi, G. COVID-19 and Italy: What next? Lancet 2020, 395, 1225–1228.
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118.
- Hulme, K.D.; Gallo, L.A.; Short, K.R. Influenza Virus and Glycemic Variability in Diabetes: A Killer Combination? Front. Microbiol. 2017, 8, 861.
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139.
- Longato, E.; Di Camillo, B.; Sparacino, G.; Saccavini, C.; Avogaro, A.; Fadini, G.P. Diabetes diagnosis from administrative claims and estimation of the true prevalence of diabetes among 4.2 million individuals of the Veneto region (North East Italy). Nutr. Metab. Cardiovasc. Dis. 2020, 30, 84–91.
- Stegenga, M.E.; van der Crabben, S.N.; Blumer, R.M.; Levi, M.; Meijers, J.C.; Serlie, M.J.; Tanck, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 2008, 112, 82–89.
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440.
- Jafar, N.; Edriss, H.; Nugent, K. The Effect of Short-Term Hyperglycemia on the Innate Immune System. Am. J. Med. Sci. 2016, 351, 201–211.
- Yan, Y.; Yang, F.; Zhu, X.; Wang, M.; Sun, Z.; Zhao, T.; Yang, X.; Zou, Y. Analysis of clinical features and pulmonary CT features of coronavirus disease 2019 (COVID-19) patients with diabetes mellitus. Endokrynol. Pol. 2020, 71, 367–375.
- Popov, D.; Simionescu, M. Alterations of lung structure in experimental diabetes, and diabetes associated with hyperlipidaemia in hamsters. Eur. Respir. J. 1997, 10, 1850–1858.
- Exley, M.A.; Hand, L.; O’Shea, D.; Lynch, L. Interplay between the immune system and adipose tissue in obesity. J. Endocrinol. 2014, 223, R41–R48.
- Polito, R.; Nigro, E.; Messina, A.; Monaco, M.L.; Monda, V.; Scudiero, O.; Cibelli, G.; Valenzano, A.; Picciocchi, E.; Zammit, C.; et al. Adiponectin and Orexin-A as a Potential Immunity Link Between Adipose Tissue and Central Nervous System. Front. Physiol. 2018, 9, 982.
- Nigro, E.; Stiuso, P.; Matera, M.G.; Monaco, M.L.; Caraglia, M.; Maniscalco, M.; Perrotta, F.; Mazzarella, G.; Daniele, A.; Bianco, A. The anti-proliferative effects of adiponectin on human lung adenocarcinoma A549 cells and oxidative stress involvement. Pulm. Pharmacol. Ther. 2019, 55, 25–30.
- Pecoraro, A.; Nigro, E.; Polito, R.; Monaco, M.L.; Scudiero, O.; Mormile, I.; Cesoni Marcelli, A.; Capasso, M.; Habetswallner, F.; Genovese, A.; et al. Total and High Molecular Weight Adiponectin Expression Is Decreased in Patients with Common Variable Immunodeficiency: Correlation with Ig Replacement Therapy. Front. Immunol. 2017, 8, 895.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9.
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448.
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8.
- Gaddam, R.R.; Chambers, S.; Bhatia, M. ACE and ACE2 in inflammation: A tale of two enzymes. Inflamm. Allergy Drug Targets 2014, 13, 224–234.
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116.
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374.
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676.
- Jones, E.S.; Vinh, A.; McCarthy, C.A.; Gaspari, T.A.; Widdop, R.E. AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol. Ther. 2008, 120, 292–316.
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427.
- Xue, D.; Li, Y.; Jiang, Z.; Deng, G.; Li, M.; Liu, X.; Wang, Y. A ROS-dependent and Caspase-3-mediated apoptosis in sheep bronchial epithelial cells in response to Mycoplasma Ovipneumoniae infections. Vet. Immunol. Immunopathol. 2017, 187, 55–63.
- Zhang, X.; Wu, M.; Jiang, H.; Hao, J.; Zhang, Q.; Zhu, Q.; Saren, G.; Zhang, Y.; Meng, X.; Yue, X. Angiotensin II upregulates endothelial lipase expression via the NF-kappa B and MAPK signaling pathways. PLoS ONE 2014, 9, e107634.
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421.
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ. Res. 2005, 97, 946–953.
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610.
- Yang, G.; Tan, Z.; Zhou, L.; Yang, M.; Peng, L.; Liu, J.; Cai, J.; Yang, R.; Han, J.; Huang, Y.; et al. Effects of Angiotensin II Receptor Blockers and ACE (Angiotensin-Converting Enzyme) Inhibitors on Virus Infection, Inflammatory Status, and Clinical Outcomes in Patients With COVID-19 and Hypertension: A Single-Center Retrospective Study. Hypertension 2020, 76, 51–58.
- Zhong, J.C.; Ye, J.Y.; Jin, H.Y.; Yu, X.; Yu, H.M.; Zhu, D.L.; Gao, P.J.; Huang, D.Y.; Shuster, M.; Loibner, H.; et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul. Pept. 2011, 166, 90–97.
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21.
- Perrotta, F.; Matera, M.G.; Cazzola, M.; Bianco, A. Severe respiratory SARS-CoV2 infection: Does ACE2 receptor matter? Respir. Med. 2020, 168, 105996.
- Ali, R.M.; Al-Shorbagy, M.Y.; Helmy, M.W.; El-Abhar, H.S. Role of Wnt4/beta-catenin, Ang II/TGFbeta, ACE2, NF-kappaB, and IL-18 in attenuating renal ischemia/reperfusion-induced injury in rats treated with Vit D and pioglitazone. Eur. J. Pharmacol. 2018, 831, 68–76.
- Zhang, W.; Li, C.; Liu, B.; Wu, R.; Zou, N.; Xu, Y.Z.; Yang, Y.Y.; Zhang, F.; Zhou, H.M.; Wan, K.Q.; et al. Pioglitazone upregulates hepatic angiotensin converting enzyme 2 expression in rats with steatohepatitis. Ann. Hepatol. 2013, 12, 892–900.
- Darwish, I.; Mubareka, S.; Liles, W.C. Immunomodulatory therapy for severe influenza. Expert. Rev. Anti Infect. Ther. 2011, 9, 807–822.
- Drucker, D.J. Coronavirus Infections and Type 2 Diabetes-Shared Pathways with Therapeutic Implications. Endocr. Rev. 2020, 41, bnaa011.
- Sestan, M.; Marinovic, S.; Kavazovic, I.; Cekinovic, D.; Wueest, S.; Turk Wensveen, T.; Brizic, I.; Jonjic, S.; Konrad, D.; Wensveen, F.M.; et al. Virus-Induced Interferon-gamma Causes Insulin Resistance in Skeletal Muscle and Derails Glycemic Control in Obesity. Immunity 2018, 49, 164–177.e6.
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841.
- van Crevel, R.; van de Vijver, S.; Moore, D.A.J. The global diabetes epidemic: What does it mean for infectious diseases in tropical countries? Lancet Diabetes Endocrinol. 2017, 5, 457–468.
- Hodgson, K.; Morris, J.; Bridson, T.; Govan, B.; Rush, C.; Ketheesan, N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology 2015, 144, 171–185.
- Boccia, M.; Aronne, L.; Celia, B.; Mazzeo, G.; Ceparano, M.; D’Agnano, V.; Parrella, R.; Valente, T.; Bianco, A.; Perrotta, F. COVID-19 and coagulative axis: Review of emerging aspects in a novel disease. Monaldi Arch. Chest Dis. 2020, 90, 271–276.
- Shah, B.R.; Hux, J.E. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care 2003, 26, 510–513.
- Muniyappa, R.; Gubbi, S. COVID-19 pandemic, coronaviruses, and diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E736–E741.
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114.
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863.
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485.
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age 2009, 31, 285–291.
- Kim, S.J.; Mehta, H.H.; Wan, J.; Kuehnemann, C.; Chen, J.; Hu, J.F.; Hoffman, A.R.; Cohen, P. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging 2018, 10, 1239–1256.
- Gu, R.; Mao, T.; Lu, Q.; Tianjiao Su, T.; Wang, J. Myeloid dysregulation and therapeutic intervention in COVID-19. Semin. Immunol. 2021, 55, 101524.
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847.


