Developmental biology is intricately regulated by epigenetics and metabolism but the mechanisms are not completely understood. The situation becomes even more complicated during diseases where all three phenomena are dysregulated. A salient example is COVID-19, where the death toll exceeded 6.96 million in 4 years, while the virus continues to mutate into different variants and infect people. Early evidence during the pandemic showed that the host’s immune and inflammatory responses to COVID-19 (like the cytokine storm) impacted the host’s metabolism, causing damage to the host’s organs and overall physiology. The involvement of angiotensin-converting enzyme 2 (ACE2), the pivotal host receptor for the SARS-CoV-2 virus, was identified and linked to epigenetic abnormalities along with other contributing factors.
- epigenetics
- metabolic reprogramming
- systemic toxicity
- diabetes mellitus
1. Introduction
2. SARS-CoV-2 Induces Metabolic Reprogramming and Epigenetic Changes
Although SARS-CoV-2 is a respiratory pathogen, its extrapulmonary involvement causes significant issues in other organ systems, such as metabolic complications and immune dysfunction, in addition to increased morbidity and mortality [10,11][9][10]. Hence, understanding the pathogenesis of extrapulmonary involvement and the resulting systemic toxicity could fill critical gaps in understanding of the disease presentation of COVID-19. A number of factors are hypothesized to contribute to systemic toxicity in COVID-19. For example, ACE2 (the functional receptor for SARS-CoV) is expressed in multiple extrapulmonary tissues [12][11] that are damaged as the result of viral infection, the subsequent inflammatory immune response with systemic cytokine release or the “cytokine storm”, which are possible mechanisms of injury [10][9]. To distinguish the pathogenesis of systemic complications of COVID-19, a study was performed where a murine model expressing the human ACE2 transgene in multiple tissues was generated, and n = 5 transgenic mice were administered SARS-CoV-2 [13][12]. As controls, a group of n = 5 transgenic mice was not administered SARS-CoV-2, and a group of n = 5 mice without the human ACE2 transgene underwent SARS-CoV-2 administration. At 7 days after systemic SARS-CoV-2 infection, mice in the experimental group developed a distinct phenotype that aligned with human COVID-19 presentation. This included severe weight loss, morbidity, neutrophilia, lymphopenia, splenic atrophy, myofibrillar disarray and myocardial edema. To better understand organ-related complications in COVID-19, organs were harvested for bulk RNA sequencing 3 days after infection (1 day prior to systemic toxicity onset) and at 7 days after infection. At 3 days post-infection, pathways related to interferon (IFN) and cytokine-mediated signaling were enriched, indicating an antiviral immune response. The response was no longer evident at 7 days post-infection, although the expression of genes regulating oxidative phosphorylation and the electron transport chain (ETC) was decreased in multiple organs. These results suggest that disease pathogenesis and the development of morbidity are associated with temporal transcription patterns [13][12]. Due to the close connections between the tricarboxylic acid cycle (TCA) and the ETC, TCA gene regulation across four organs was examined in a study. The downregulation of the TCA cycle genes was consistent across the heart, lung, kidney and spleen. In line with these observations, metabolomic profiling confirmed the lower TCA cycle metabolite levels in the serum of the experimental group. Finally, DNA methylation analysis of the heart and kidney at 7 days post-infection was performed to investigate whether epigenetic changes could contribute to gene expression in multiple organs. This revealed differentially methylated sites in the heart (172 sites) and kidney (49 sites), suggesting that tissue-specific epigenetic changes occur soon after SARS-CoV-2 infection [13][12]. Other studies have also reported the adverse effects of COVID-19 on the heart [14][13].3. COVID-19 Is Associated with Accelerated Epigenetic Aging and Hence Development
Chronological age is an established and independent [15][14] risk factor of severity and death in COVID-19 patients [16,17][15][16]. Additionally, epigenetic studies have indicated that markers of biological age, like DNA methylation (DNAm), are associated with severe COVID-19 [18][17]. The disparity between biological and chronological age (epigenetic age acceleration) has been related to survival outcomes in age-related diseases [19][18]. However, little is known about epigenetic aging during severe vs. non-severe COVID-19, and whether it could predict disease severity and model disease progression. To assess the association between accelerated epigenetic aging and SARS-CoV-2 infection severity, a genome-wide DNA methylation study was conducted on the whole blood samples of 232 healthy individuals, 194 individuals with non-severe COVID-19 and 213 individuals with severe COVID-19 [5]. The epigenetic age acceleration of individuals was calculated by determining the difference between their chronological age and epigenetic age (calculated by applying epigenetic clocks and telomere length estimators (DNAmTL) to an individual’s methylation profile). Individuals with severe COVID-19 showed significant DNAm age acceleration and DNAmTL attrition acceleration compared to individuals with non-severe COVID-19. In comparison, individuals with non-severe COVID-19 had significantly higher DNAm and DNAmTL acceleration compared to healthy individuals [5]. To further understand these associations, a study analyzed the dynamic acceleration of epigenetic aging in six individuals with COVID-19 and six uninfected controls across different disease phases, defined by inflammatory markers and temporal disease severity [20][19]. Increasing age acceleration was observed in the initial disease phases, which could then be partially reversed in the later phases, although the mechanism behind this observation requires further research.4. Epigenetic Regulation of Viral Pathogenicity Suggests Epi-Drugs as a Therapeutic Approach against COVID-19
During infection, SARS-CoV-2 uses transmembrane serine protease 2 (TMPRSS2) and the ACE2 receptor to infect the host cell [22,23][20][21] and the RNA polymerase to synthesize viral proteins [24][22]. As a response, the host immune system causes a “cytokine storm”, which can lead to an uncontrolled inflammatory response [25][23], culminating in lung injury, acute respiratory distress syndrome and organ failure. Epigenetic alterations such as DNA methylation have crucial roles in most biological processes and modify genetic expression to allow cells to adapt to environmental changes [26][24]. For example, epigenetic pathways could impact the expression of genes like ACE2 and immunoregulatory genes involved throughout COVID-19’s pathogenesis [26,27][24][25]. Epigenetic pathways may be altered by SARS-CoV-2 and are also linked to COVID-19 severity [28][26]. Hence, further insights into epigenetics would allow the possibility of more precise therapies against COVID-19. The overexpression of ACE2 is associated with higher COVID-19 severity [29][27], and epigenetic processes are responsible for ACE2 control and expression [26][24]. Histone deacetylase (HDAC) can also the modulate epigenetic effects on COVID-19: HDAC upregulates ACE2 expression, which promotes viral entry into cells [26][24]. It also activates proinflammatory responses against viral infection, which can contribute to the cytokine storm [26][24]. Conversely, histone deacetylase inhibitors have been reported to downregulate ACE2 and the production of infectious SARS-CoV-2 [26,30][24][28], suggesting a potential therapy for COVID-19 (Figure 1). The cytokine storm, which causes many of the adverse health outcomes in COVID-19, could also potentially be regulated through epigenetic modulation [31][29].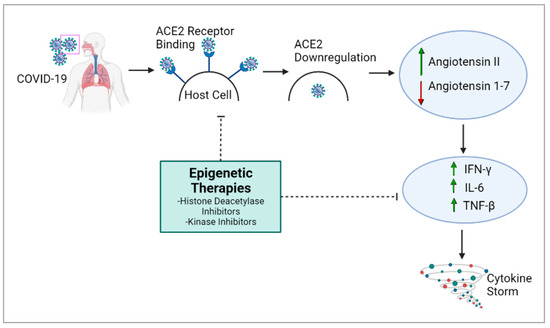
5. Epigenetic Therapies May Help to Mitigate COVID-19 Severity
Modulation of the epigenetic landscape largely determines differential gene expression in several diseases. Several key genes involved in COVID-19 are impacted by epigenetic pathways [32][30]. Hence, further research into these pathways would improve understanding of the disease and epigenetic therapies could be used to mitigate COVID-19. For COVID-19 therapies, the major target genes would be ACE2, TMPRSS2 and Furin to prevent the entry of the virus and cytokines. As mentioned earlier, ACE2 is the receptor for SARS-CoV-2. TMPRSS2 and Furin also trigger SARS-CoV-2 infection by cleaving ACE2, which promotes viral uptake and allows cell entry [32][30]. ACE2 gene expression is downregulated by DNA methylation and histone modification, offering a mechanism for therapy [32][30]. For example, histone methyltransferase EZH2-mediated H3K27me3 modifications of the ACE2 promoter could be a target for COVID-19 therapies [33][31]. In vitro data suggest that vitamin D and quercetin could inhibit ACE2 and Furin, thereby mitigating COVID-19’s severity [34,35][32][33]. However, the prevalence of ACE2’s function in physiology, especially in the cardiovascular and renal systems, renders ACE2 inhibitors risky in a clinical setting [36,37][34][35].6. Vitamin D Has a Plausible Protective Effect against COVID-19
The vitamin D endocrine system regulates 3% of the human genome, and it is heavily involved in both innate and adaptive immunity [34][32]. Active vitamin D is crucial for immune regulation, while its deficiency has been associated with chronic lung diseases [41][36]. In the airway epithelium, vitamin D controls vitamin D receptor (VDR)-induced gene expression and eliminates pathogens via CD14, antimicrobial peptide mechanisms and the promotion of autophagy [34][32]. An analysis of 20 patients hospitalized with COVID-19 indicated that 75% had a vitamin D deficiency [42][37]. Additionally, a study of 43 individuals reported that a combination of vitamin D, magnesium and vitamin B12 was associated with a significant reduction in the proportion of patients who deteriorated [43][38]. Although the immune functions of vitamin D are known, well-designed trials are necessary to establish a plausible protective role of vitamin D in COVID-19.7. Overview of Metabolic Abnormalities Associated with COVID-19
In addition to epigenetic disruptions, COVID-19 results in metabolic aberrations that can alter the energy metabolism and cause changes in appetite, with the body burning more calories to support the elevated immune response. COVID-19 has been associated with increased insulin resistance, which can negatively impact glucose metabolism [44][39]. This can lead to changes in blood sugar levels and an increased risk of type 2 diabetes. The immune response to COVID-19 can cause inflammation, which can affect various metabolic processes. Chronic inflammation has also been linked to various health conditions, such as obesity, metabolic syndrome and cardiovascular disease. One of the indirect effects of COVID-19 on metabolism is weight gain. With many people leading sedentary lifestyles and work routines, especially during the 2+ years of the COVID-19 pandemic, there has been a marked increase in excessive calorie intake and a lack of sufficient physical activity to burn these calories. This dangerous combination has led to weight gain and associated illnesses worldwide. Metabolic and vascular problems were present in up to 50% of COVID-19 fatalities [45][40]. Furthermore, COVID-19 and the metabolic and endocrine systems have several direct connections. As a result, individuals with metabolic dysfunction (such as obesity, hypertension, non-alcoholic fatty liver disease and diabetes) are not only more likely to develop severe COVID-19, but a SARS-CoV-2 infection may also bring about new cases of diabetes or worsen pre-existing metabolic diseases. COVID-19, in conjunction with type 2 diabetes and obesity, which are both characterized by severe insulin resistance [46][41], has numerous consequences. Almost 4 years have passed since the initial outbreak of SARS-CoV-2. Research during this period has revealed that people with metabolic diseases are not only more susceptible to severe COVID-19, but also have an increased risk of post-acute sequelae of COVID-19 and vaccine breakthroughs [47,48,49][42][43][44]. To address these concerns, high-throughput omics-based research on large cohort studies using samples from COVID-19 patients has shed significant light on its etiology, prognosis and outcomes. This has established a new specialization called COVIDomics, which encompasses omics-level research on COVID-19 diagnosis, prevention and biomarkers; the identification of therapeutic targets; and all other aspects associated with SARS-CoV-2 infection [50][45]. COVIDomics has been explored in a review by Costanzo et al., with a very robust and comprehensive analysis of metabolomics, lipidomics and proteomics studies on plasma, serum and infected cells from COVID-19 patients and a multiomics integrational analysis [50][45].8. Abnormal Metabolism and Diabetes Are Often Manifested in COVID-19
Several studies present associations between COVID-19 severity, increased mortality, diabetes mellitus and the individual degree of hyperglycemia [51,52,53,54,55,56][46][47][48][49][50][51]. The development of temporary insulin resistance in adults with type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM) has been linked to acute respiratory viral infections like COVID-19, and hyperglycemia has also been linked with severe COVID-19 [56][51]. Based on a commonly accepted explanation, these individuals are predisposed to the excessive release of cytokines, or a “cytokine storm”, since they experience a state of chronic metabolic inflammation. These elevated levels of inflammatory cytokines might in turn trigger multi-organ failure [56][51]. The main entry receptor for SARS-CoV-2 is angiotensin-converting enzyme 2 (ACE2). The ability of the pancreas to produce insulin in response to hyperglycemia may be impaired when SARS-CoV-2 binds to pancreatic ACE2 receptors, causing damage to the islets [56][51]. Many other pathophysiological processes have also been suggested, such as elevated levels of tissue-associated enzymes, the altered expression of ACE2 receptors, immune regulatory dysregulation, pulmonary and endothelium dysfunction, systemic inflammation and hypercoagulability and higher concentrations of anti-inflammatory biomarkers including IL-6, D-dimers and C-reactive protein. All these pathophysiological issues may contribute to an increased response to the cytokine storm that causes inflammation in patients with T1DM or T2DM, which may lead to a more severe COVID-19 course [56][51]. Additionally, a 2020 analysis of eight cohort studies indicated that COVID-19 patients with excess adiposity had a higher risk of death and serious illness [57][52]. Obesity and low-grade systemic inflammation are common in individuals with cardiometabolic disorders, and this may be a possible pathway connecting severe COVID-19 with insulin resistance, hypertension, cardiovascular disease and T2DM. The chronic care of patients with T2DM can result in fewer microvascular and macrovascular problems when risk variables such blood pressure, dyslipidemia and glucose levels are managed [58][53]. There is proof that multifactorial risk factor therapies have a lasting positive impact on mortality, cardiovascular and renal outcomes [58][53]. The major cause of death from COVID-19 is acute respiratory distress syndrome (ARDS), which develops as a result of an accelerated inflammatory response that releases proinflammatory cytokines including interleukin (IL) and tumor necrosis factor alpha [59][54]. The family of proteins known as Toll-like receptors (TLRs) serves as sensors and aids the immune system in distinguishing between its own cells and invaders. In the host cell membrane, SARS-CoV-1 and, most likely, SARS-CoV-2 interact with TLR to promote the expression of the main response gene for myeloid 88 (MyD88) differentiation [60][55]. This then causes nuclear factor kappa beta to become active, ultimately triggering an inflammatory cascade that worsens lung injury [61][56].9. Hyperglycemia Is Associated with COVID-19 Severity
Chronic hyperglycemia is a condition that impairs both antibody-mediated immunity and innate immunity. Chronic low-grade inflammation may contribute to diabetes by inducing insulin resistance, disrupting glucose control and elevating inflammatory markers [62,63,64][57][58][59]. An increase in IL-6 and C-reactive protein (RCP) levels was observed in diabetic individuals who had the SARS-CoV-2 virus. This proinflammatory state of diabetes led to the release of cytokines and a systemic inflammatory response that accompanied acute respiratory distress syndrome (ARDS) in COVID-19 patients [65,66][60][61]. Many of the mechanisms that connect diabetes and hypertension interact in bidirectional ways. First, this complex network is affected by several typical biological factors, including renin–angiotensin–aldosterone system (RAAS) homeostasis, elevated oxidative stress, systemic proinflammatory conditions and enhanced sympathetic nervous system (SNS) activation [67][62]. The activity of insulin-mediated vasodilator and vasoconstrictor molecules is unbalanced as a result of decreased insulin sensitivity. This results in the vascular system’s remodeling, stiffening and fibrosis, which are predominantly controlled by MAPK-dependent signaling pathways. In fact, insulin increases the production of several vasoconstrictor mediators such as vascular cell adhesion molecule 1, PAI-1 and endothelin-1 [68,69,70][63][64][65]. Additionally, diabetes and the hypertension risk and severity are influenced by obesity and adipose tissue hormone release [71][66]. Similar to hypertension, excess body fat may change how the pulmonary viral pathogenesis, milieu and immune cell trafficking interact [67,72][62][67]. The relationships between many organs and metabolic processes are complicated, but the three primary ones that appear to be at play are inflammation, ACE-2 receptor modulation and hyperglycemia and immune system dysregulation. In patients with chronic SARS-CoV-2 infection, hyperglycemia has been linked to both illness severity and mortality [71][66]. These results are remarkably in line with research on patients with highly virulent avian influenza, SARS and MERS, where uncontrolled hyperglycemia was linked to worse outcomes. Numerous biochemical pathways, such as a changed immunological response, an inflammatory echo and the modulation of the virus’s receptor expression utilized for cell entrance, have been hypothesized as links between hyperglycemia and SARS-CoV-2 infection. In fact, elevated blood sugar levels may promote viral entrance and replication in vivo, potentially by changing the ACE2 receptor [72,73,74][67][68][69]. Increased glucose levels may also inhibit the immune system’s ability to fight viruses, making viral infections more severe. Reduced neutrophil degranulation, phagocytic activity and chemotaxis inhibit the lymphocyte proliferative response and impair the complement activation of the innate and adaptive immune responses, all of which are affected by hyperglycemia [75,76][70][71]. It is believed that diabetes-related hyperglycemia impairs the immune system’s ability to prevent the spread of infection in diabetic people [76][71]. According to a study, hyperglycemia significantly lowers the macrophagic activity of both macrophages and neutrophils, leaving patients more vulnerable to infection [77][72]. High glucose levels are associated with reduced vascular dilatation and increased permeability during the early stages of inflammatory reactions, potentially as a result of protein kinase C activation [78][73]. Additionally, hyperglycemia can directly glycolyze proteins and affect the tertiary structure of complements. These modifications reduce phagocytosis and impair the immunoglobulin-mediated opsonization of bacteria, as well as complement fixation to bacteria [78][73]. As a result, the evidence points to a dysregulated immune response as the likely cause of the higher disease severity seen in people with SARS-CoV-2 infection and associated hyperglycemia. This leads to a more severe and extended pathology. A substantial link between hyperglycemia and a worsened result from SARS-CoV-2 has also been demonstrated. COVID-19 individuals with T2DM are reported to be more prone to having increased inflammatory markers, hypercoagulability and abnormalities of glucose and lipid metabolism (Figure 2). They are also more likely to have hypoproteinemia [79][74].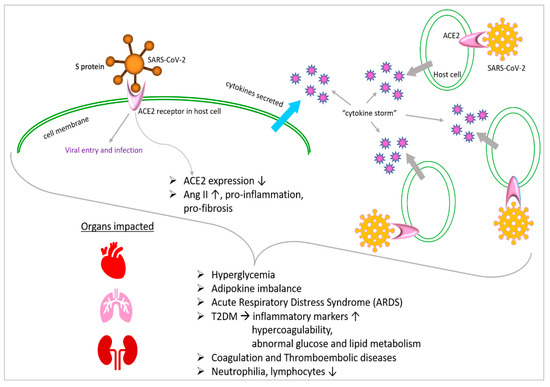
10. COVID-19 Affects Adipokines with Impacts on Glucose Metabolism
In order to effectively regulate the clinical outcomes in infected patients with coexisting obesity, hyperglycemia and diabetes, a glycemic profile must be established in SARS-CoV-2 patients. As previously mentioned, obesity may be another relevant factor linked to a poor prognosis in COVID-19 patients. Adipose tissue and the immune system’s intricate communication may be significant to SARS-CoV-2 infection. Although immune cells are present throughout the adipose tissue in the normal state, adipocytes and immune cells are in a state of equilibrium, which in turn results in the synthesis of adipokines [80][75]. A large number of immune cells invade the adipose tissue in pathophysiological circumstances like obesity, which causes an imbalance in the synthesis of adipokines, including leptin and adiponectin [81,82][76][77]. Additionally, via raising insulin sensitivity and glucose uptake and thus raising GLUT-4 translocation, these adipokines are implicated in the glucose balance, energy homeostasis and insulin sensitivity [81,83][76][78].11. COVID-19 Affects Metabolism through Interactions with Angiotensin
A spike (S) protein that protrudes from the viral envelope is responsible for the attachment and adherence of coronaviruses to host or human cells [53,84][48][79]. It has been confirmed that the S1 (subunit of the SARS-CoV-2 spike protein) region of SARS-CoV and SARS-CoV-2 binds to angiotensin-converting enzyme 2 (Figure 1). This interaction occurs through the receptor-binding domain (RBD) of S1. The mono-carboxypeptidase ACE2 was first discovered as an ACE receptor homolog in 2000 [85[80][81],86], and, since then, its molecular structure has been completely characterized [87][82]. A wide range of biological systems, including bladder urothelial cells, kidney proximal tubule cells, cholangiocytes, enterocytes, esophageal epithelial cells, myocardial cells and type II lung alveolar cells, express ACE2 [88][83]. By cleaving a single amino acid in the human lung, ACE2 produces angiotensin (I–VII) from angiotensin II [89][84]. Ang (angiotensin)-(I–VII), through Mas receptor (Mas1) activation, is expressed on endothelial cells and results in vasodilatory, anti-inflammatory and antifibrotic effects [90][85]. It is interesting to note that the interaction between SARS-CoV-2 and ACE2 causes ACE2 expression to be downregulated, which causes the aggregation of AngII (angiotensin II) with proinflammatory and profibrotic effects [91,92][86][87]. In a limited cohort study, plasma samples infected with SARS-CoV-2 were shown to have significantly higher levels of Ang II [93][88]. Apoptotic stimuli could be activated by many signaling pathways. First, it has been discovered that elevated oxidative stress, which is caused by a hyperactive AngII/AT1R/NAPDHox axis, is linked to cardiovascular diseases, including hypertension and atherosclerosis [94,95][89][90]. Hence, the production of reactive oxygen species (ROS) downstream triggers the release of CytC from damaged mitochondria [96][91], the p38MAPK/JNK cascade or the activation of caspase 3 [97][92], all of which are known to induce apoptosis. Additionally, proapoptotic signals have also been linked to nuclear factor kappa B (NF-kb) activation and the production of cytokines such as interleukin-6, IL-1 and tumor necrosis factor alpha (TNFα) [98][93]. When cyclo-oxygenase 2 (COX2) is elevated due to high levels of AngII and proinflammatory cytokines, ROS and inflammatory prostaglandin E2 are produced as a result [99][94].12. Metabolism-Related Therapeutics Could Be Promising against COVID-19
It is interesting to note that polymorphisms for ACE2 were independently linked to a higher risk of developing hypertension and cardiovascular problems in people with diabetes [100,101][95][96]. In the late stage of SARS-CoV-2 infection, the overactivation of these pathways can lead to a condition of hyperinflammation. Agents that operate on the renin–angiotensin system (RAS) noticeably alter ACE2 expression. The foundation of many therapies for cardiovascular and renal illnesses is the use of RAS inhibitors. To stop diabetic nephropathy and cardiovascular remodeling, these medicines are frequently prescribed to diabetic patients. Different responses to the administration of agents that interfere with this regulatory axis have been observed in experimental and clinical models. In particular, angiotensin II receptor blockers (ARBs) and mineralocorticoid receptor blockers appear to increase the levels of ACE2 expression [102[97][98],103], whereas the administration of ACE inhibitors, which increased cardiac ACE2 mRNA levels, did not result in higher ACE2 activity [103][98]. In a different study, olmesartan-treated hypertension patients had higher urine ACE2 levels [104][99]. However, several international societies and associations have advised against stopping angiotensin-converting enzyme inhibitors (ACE-Is) or sartans in patients receiving long-term therapy, based on the previously mentioned mechanisms connecting ACE2 expression with local antiproliferative, antifibrotic and anti-inflammatory properties [105][100]. Other diabetes medications may inhibit the RAS’s ability to function normally. An important family of insulin sensitizers used in the treatment of T2DM is thiazolidinediones. The modification of peroxisome proliferator-activated receptors mediates the molecular processes of the biological reactions to thiazolidinediones in diabetic patients (PPARs). Thiazolidinediones, like ACEIs and ARBs, increase ACE2 expression [106[101][102],107], which could expose alveolar cells to SARS-CoV-2 infection. PPARs are inflammatory mediators with possible immunoregulatory properties. The inflammatory cytokines IL-6 and INF, which are heavily connected in SARS-CoV-2, are reduced as a result of their activation [108][103]. Insulin-mimetic medications like rosiglitazone and pioglitazone, which are used to treat T2DM, have a significant overall ability to reduce influenza virus infection [108][103]. The cardiovascular and renal advantages of sodium-glucose cotransporter 2 inhibitor (SGLT2) therapy for those with T2DM are now also established, being applied those without T2DM, according to studies [58][53]. Given the pathophysiology of COVID-19 and the benefits of SGLT2 inhibitors and glucagon-like peptide-1 receptor agonists (GLP1RA) that have been documented, these treatments may be preferable to other therapeutic options for patients with T2DM and long COVID, and maybe even for those without diabetes mellitus. Therapeutic interventions towards metabolism are implemented in several clinical trials against COVID-19, e.g., NCT04517396, NCT04542213 and NCT04573764 (Figure 3 and Table 1).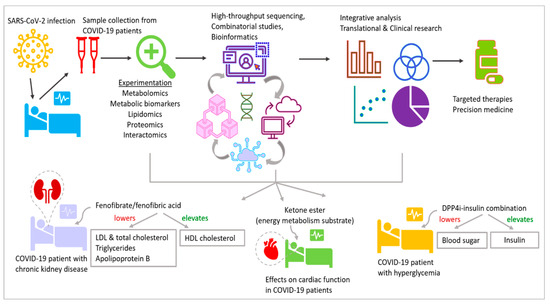
13. Inflammatory Immune Response in Diabetic COVID-19 Patients Is Deleterious
It has been established that infectious diseases significantly increase mortality in diabetes patients by linking acute viral respiratory infection to the fast development of transitory insulin resistance in both overweight individuals and healthy euglycemic normal-weight individuals [109,110][104][105]. Older diabetes patients have higher death rates, according to a retrospective review [111][106]. Due to a combination of dysregulated innate immunity and inflammatory responses, diabetes is closely linked to an elevated risk and worse outcomes for bacterial and viral infections [112,113][107][108]. The gastrointestinal tract and impaired epithelial barrier function in the lungs of diabetic people also make those with coronavirus infection susceptible to subsequent bacterial infections [109][104]. The death of patients affected by SARS-CoV-2 is a result of chronic inflammation caused by the synthesis of related cytokines during viral infection. These conditions include coagulation activation, neutrophilia and kidney damage [65,66,114][60][61][109]. Several studies have shown that, in diabetic patients with SARS-CoV-2, the absolute count of lymphocytes in the peripheral blood is significantly lower, while the absolute count of neutrophils is remarkably higher [115][110]. Additionally, compared to non-diabetic patients with SARS-CoV-2, diabetic individuals had increased serum levels of inflammatory-related biomarkers [80][75]. These people are distinguished particularly by increased blood levels of IL-6, TNF-α, C-reactive protein (CRP) and serum ferritin. Among them, IL-6 has a longer expression duration than other cytokines (IL-1 and TNF-α) and is a predictor of the severity of illness and prognosis [116][111]. Additionally, it is also observed that there is an increase in serum ferritin in diabetes patients, confirming the activation of the monocyte–macrophage system, an essential component of the inflammatory storm [65,66,116][60][61][111].14. Immunometabolic Phenotyping Reveals T Cell and Myeloid Cell Populations Unique to Severe COVID-19
The factors that determine why some patients quickly recover from SARS-CoV-2 infection while others experience severe disease, potentially leading to death, remain unclear. Previous research has identified associations between COVID-19 and generalized changes in immune cell subsets [117][112]. However, these studies do not consider host deficiencies specific to SARS-CoV-2 as opposed to other viral infections [118][113]. Furthermore, investigating the reciprocal interaction between metabolic reprogramming and immune function in the context of COVID-19 pathogenesis could provide novel insights. To examine metabolic programs at the single-cell level, a flow-cytometry-based proteomic and epigenetic approach was utilized. Samples of peripheral blood mononuclear cells (PBMCs) from individuals with acute COVID-19 and recovered individuals were compared to those of healthy controls, individuals hospitalized with influenza, individuals with acute hepatitis C and individuals with chronic hepatitis C. In an unbiased analysis of T cells employing combined immune and metabolic markers, a population of T cells unique to patients with acute COVID-19 was identified. This population exhibited the upregulation of voltage-dependent anion channel 1 (VDAC1)—a mitochondrial membrane protein involved in metabolite transport and mitochondrial cell death signaling [119][114]—and the upregulation of histone H3 lysine 27 trimethylation (H3K27me3), which is an epigenetic modification for transcription repression [118][113]. Interestingly, this subset of cells also expresses low levels of the glucose transporter 1 and hexokinase II (HKII) proteins, which are typically upregulated alongside VDAC1 and H3K27me3 [6], and higher levels of translocase of outer mitochondrial membrane 20 (Tomm20) and killer cell lectin-like receptor subfamily G member 1 (KLRG1, associated with T cell senescence) [120,121][115][116]. To determine whether elevated VDAC1 and Tomm20 indicate altered mitochondrial function, electron microscopy was performed on PBMCs from acute COVID-19 patients and healthy controls. In severe COVID-19 patients, the mitochondria were irregularly shaped, and cytochrome c was found in the cytosol (subsequently inducing apoptosis), suggesting that high VDAC1 expression makes these T cells more susceptible to death. However, this apoptosis was inhibited in vitro by targeting VDAC1 oligomerization. The study also found that the prevalence of the unique T cells increased with age (an established risk factor for severe COVID-19). These results suggest that targeting mitochondrial dysfunction could be a possible course for treatment. The study also used an immunometabolic assay to examine myeloid cells in PBMC samples, which showed increased inflammation and activation in COVID-19 patients in previous studies [122][117]. Myeloid-derived suppressor cells with metabolic phenotypes unique to severe COVID-19 were identified. The frequency of these cells was positively associated with the COVID-19 severity, suggesting that they may indicate dysregulated inflammation. These results provide more cell-specific insights into the factors associated with COVID-19’s severity and distinguish subsets of immune cells that could help to predict disease severity and be used as metabolic targets for future treatment.15. Hyperglycemia Associated with COVID-19 Impacts Blood Coagulation
Hyperglycemia is a significant risk factor for a defective coagulation balance and platelet aggregation, which may contribute to the worsened thromboembolic diseases seen in deceased COVID-19 individuals. In diabetic patients, a number of mechanisms have been investigated that connect inflammation with coagulative homeostasis [114][109]. First, inflammation activates plasmin, which increases D-dimer. Second, severe inflammation and hypoxia activate monocyte–macrophages, and thrombin activation results in the secretion of a large number of tissue factors and the activation of the exogenous coagulation pathway, which results in a general hypercoagulable state or even disseminated intravascular coagulation [65,66,114][60][61][109]. Furthermore, increased D-dimer levels are frequently observed, and they show a steady increase when the disease begins to worsen [123][118]. Thus, the longitudinal analysis of lymphocyte count dynamics and inflammatory markers like IL-6, CRP and ferritin over the course of the disease may assist in the detection of people with a poor prognosis and the initiation of prompt treatment to improve outcomes.References
- Spinney, L. How pandemics shape social evolution. Nature 2019, 574, 324–326.
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160.
- Prajapati, J.; Rao, P.; Poojara, L.; Goswami, D.; Acharya, D.; Patel, S.K.; Rawal, R.M. Unravelling the antifungal mode of action of curcumin by potential inhibition of CYP51B: A computational study validated in vitro on mucormycosis agent, Rhizopus oryzae. Arch. Biochem. Biophys. 2021, 712, 109048.
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298.
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135.
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther. Nucleic Acids 2019, 17, 24–37.
- Santorelli, L.; Caterino, M.; Costanzo, M. Dynamic Interactomics by Cross-Linking Mass Spectrometry: Mapping the Daily Cell Life in Postgenomic Era. OMICS 2022, 26, 633–649.
- Mar, D.; Babenko, I.M.; Zhang, R.; Noble, W.S.; Denisenko, O.; Vaisar, T.; Bomsztyk, K. A High-Throughput PIXUL-Matrix-Based Toolbox to Profile Frozen and Formalin-Fixed Paraffin-Embedded Tissues Multiomes. Lab. Investig. 2024, 104, 100282.
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032.
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol. 2020, 5, 751–753.
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637.
- Li, S.; Ma, F.; Yokota, T.; Garcia, G., Jr.; Palermo, A.; Wang, Y.; Farrell, C.; Wang, Y.C.; Wu, R.; Zhou, Z.; et al. Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2-induced systemic toxicity. JCI Insight 2021, 6, r145027.
- Sarkar, S.; Sen, R. Insights into Cardiovascular Defects and Cardiac Epigenome in the Context of COVID-19. Epigenomes 2022, 6, 13.
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981.
- Levin, A.T.; Hanage, W.P.; Owusu-Boaitey, N.; Cochran, K.B.; Walsh, S.P.; Meyerowitz-Katz, G. Assessing the age specificity of infection fatality rates for COVID-19: Systematic review, meta-analysis, and public policy implications. Eur. J. Epidemiol. 2020, 35, 1123–1138.
- Gupta, S.; Hayek, S.S.; Wang, W.; Chan, L.; Mathews, K.S.; Melamed, M.L.; Brenner, S.K.; Leonberg-Yoo, A.; Schenck, E.J.; Radbel, J.; et al. Factors Associated With Death in Critically Ill Patients With Coronavirus Disease 2019 in the US. JAMA Intern. Med. 2020, 180, 1436–1447.
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26.
- Yu, M.; Hazelton, W.D.; Luebeck, G.E.; Grady, W.M. Epigenetic Aging: More Than Just a Clock When It Comes to Cancer. Cancer Res. 2020, 80, 367–374.
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314.e9.
- Mollica, V.; Rizzo, A.; Massari, F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol 2020, 16, 2029–2033.
- Beyerstedt, S.; Casaro, E.B.; Rangel, E.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170.
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708.
- Dey, A.; Vaishak, K.; Deka, D.; Radhakrishnan, A.K.; Paul, S.; Shanmugam, P.; Daniel, A.P.; Pathak, S.; Duttaroy, A.K.; Banerjee, A. Epigenetic perspectives associated with COVID-19 infection and related cytokine storm: An updated review. Infection 2023, 51, 1603–1618.
- Kgatle, M.M.; Lawal, I.O.; Mashabela, G.; Boshomane, T.M.G.; Koatale, P.C.; Mahasha, P.W.; Ndlovu, H.; Vorster, M.; Rodrigues, H.G.; Zeevaart, J.R.; et al. COVID-19 Is a Multi-Organ Aggressor: Epigenetic and Clinical Marks. Front. Immunol. 2021, 12, 752380.
- AbdelHamid, S.G.; Refaat, A.A.; Benjamin, A.M.; Elmawardy, L.A.; Elgendy, L.A.; Manolly, M.M.; Elmaksoud, N.A.; Sherif, N.; Hamdy, N.M. Deciphering epigenetic(s) role in modulating susceptibility to and severity of COVID-19 infection and/or outcome: A systematic rapid review. Environ. Sci. Pollut. Res. Int. 2021, 28, 54209–54221.
- Gracia-Ramos, A.E. Is the ACE2 Overexpression a Risk Factor for COVID-19 Infection? Arch. Med. Res. 2020, 51, 345–346.
- Takahashi, Y.; Hayakawa, A.; Sano, R.; Fukuda, H.; Harada, M.; Kubo, R.; Okawa, T.; Kominato, Y. Histone deacetylase inhibitors suppress ACE2 and ABO simultaneously, suggesting a preventive potential against COVID-19. Sci. Rep. 2021, 11, 3379.
- Yasmin, R.; Siraj, S.; Hassan, A.; Khan, A.R.; Abbasi, R.; Ahmad, N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediat. Inflamm. 2015, 2015, 201703.
- Patra, S.K.; Szyf, M. Epigenetic perspectives of COVID-19: Virus infection to disease progression and therapeutic control. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166527.
- Li, Y.; Li, H.; Zhou, L. EZH2-mediated H3K27me3 inhibits ACE2 expression. Biochem. Biophys. Res. Commun. 2020, 526, 947–952.
- Foolchand, A.; Mazaleni, S.; Ghazi, T.; Chuturgoon, A.A. A Review: Highlighting the Links between Epigenetics, COVID-19 Infection, and Vitamin D. Int. J. Mol. Sci. 2022, 23, 12292.
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andreo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822.
- de Abajo, F.J.; Rodriguez-Martin, S.; Lerma, V.; Mejia-Abril, G.; Aguilar, M.; Garcia-Luque, A.; Laredo, L.; Laosa, O.; Centeno-Soto, G.A.; Angeles Galvez, M.; et al. Use of renin-angiotensin-aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: A case-population study. Lancet 2020, 395, 1705–1714.
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507.
- Hughes, D.A.; Norton, R. Vitamin D and respiratory health. Clin. Exp. Immunol. 2009, 158, 20–25.
- Lau, F.H.; Majumder, R.; Torabi, R.; Saeg, F.; Hoffman, R.; Cirillo, J.D.; Greiffenstein, P. Vitamin D insufficiency is prevalent in severe COVID-19. medRxiv 2020.
- Tan, C.W.; Ho, L.P.; Kalimuddin, S.; Cherng, B.P.Z.; Teh, Y.E.; Thien, S.Y.; Wong, H.M.; Tern, P.J.W.; Chandran, M.; Chay, J.W.M.; et al. Cohort study to evaluate the effect of vitamin D, magnesium, and vitamin B(12) in combination on progression to severe outcomes in older patients with coronavirus (COVID-19). Nutrition 2020, 79–80, 111017.
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785.
- Stefan, N. Metabolic disorders, COVID-19 and vaccine-breakthrough infections. Nat. Rev. Endocrinol. 2022, 18, 75–76.
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B. Global pandemics interconnected—Obesity, impaired metabolic health and COVID-19. Nat. Rev. Endocrinol. 2021, 17, 135–149.
- Brosh-Nissimov, T.; Orenbuch-Harroch, E.; Chowers, M.; Elbaz, M.; Nesher, L.; Stein, M.; Maor, Y.; Cohen, R.; Hussein, K.; Weinberger, M.; et al. BNT162b2 vaccine breakthrough: Clinical characteristics of 152 fully vaccinated hospitalized COVID-19 patients in Israel. Clin. Microbiol. Infect. 2021, 27, 1652–1657.
- Maestre-Muniz, M.M.; Arias, A.; Mata-Vazquez, E.; Martin-Toledano, M.; Lopez-Larramona, G.; Ruiz-Chicote, A.M.; Nieto-Sandoval, B.; Lucendo, A.J. Long-Term Outcomes of Patients with Coronavirus Disease 2019 at One Year after Hospital Discharge. J. Clin. Med. 2021, 10, 2945.
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the Mystery Surrounding Post-Acute Sequelae of COVID-19. Front. Immunol. 2021, 12, 686029.
- Costanzo, M.; Caterino, M.; Fedele, R.; Cevenini, A.; Pontillo, M.; Barra, L.; Ruoppolo, M. COVIDomics: The Proteomic and Metabolomic Signatures of COVID-19. Int. J. Mol. Sci. 2022, 23, 2414.
- Holman, N.; Knighton, P.; Kar, P.; O’Keefe, J.; Curley, M.; Weaver, A.; Barron, E.; Bakhai, C.; Khunti, K.; Wareham, N.J.; et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 823–833.
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581.
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95.
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e1063.
- Barron, E.; Bakhai, C.; Kar, P.; Weaver, A.; Bradley, D.; Ismail, H.; Knighton, P.; Holman, N.; Khunti, K.; Sattar, N.; et al. Associations of type 1 and type 2 diabetes with COVID-19-related mortality in England: A whole-population study. Lancet Diabetes Endocrinol. 2020, 8, 813–822.
- Lim, S.; Bae, J.H.; Kwon, H.S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30.
- Seidu, S.; Gillies, C.; Zaccardi, F.; Kunutsor, S.K.; Hartmann-Boyce, J.; Yates, T.; Singh, A.K.; Davies, M.J.; Khunti, K. The impact of obesity on severe disease and mortality in people with SARS-CoV-2: A systematic review and meta-analysis. Endocrinol. Diabetes Metab. 2021, 4, e00176.
- Khunti, K.; Kosiborod, M.; Ray, K.K. Legacy benefits of blood glucose, blood pressure and lipid control in individuals with diabetes and cardiovascular disease: Time to overcome multifactorial therapeutic inertia? Diabetes Obes. Metab. 2018, 20, 1337–1341.
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720.
- Mabrey, F.L.; Morrell, E.D.; Wurfel, M.M. TLRs in COVID-19: How they drive immunopathology and the rationale for modulation. Innate Immun. 2021, 27, 503–513.
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schafer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15.
- Iacobellis, G. COVID-19 and diabetes: Can DPP4 inhibition play a role? Diabetes Res. Clin. Pract. 2020, 162, 108125.
- Kokic Males, V. Letter to the editor in response to the article “COVID-19 and diabetes: Can DPP4 inhibition play a role?”. Diabetes Res. Clin. Pract. 2020, 163, 108163.
- Morin, N. Response to COVID-19 and diabetes: Can DPP4 inhibition play a role?—GLP-1 might play one too. Diabetes Res. Clin. Pract. 2020, 164, 108160.
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3319.
- Blanke, C.D. In response: Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, 36, e3331.
- Fadini, G.P.; Morieri, M.L.; Longato, E.; Avogaro, A. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J. Endocrinol. Investig. 2020, 43, 867–869.
- Sun, Y.; Wang, Q.; Yang, G.; Lin, C.; Zhang, Y.; Yang, P. Weight and prognosis for influenza A(H1N1)pdm09 infection during the pandemic period between 2009 and 2011: A systematic review of observational studies with meta-analysis. Infect. Dis. 2016, 48, 813–822.
- Carter, S.J.; Baranauskas, M.N.; Fly, A.D. Considerations for Obesity, Vitamin D, and Physical Activity Amid the COVID-19 Pandemic. Obesity 2020, 28, 1176–1177.
- Remuzzi, A.; Remuzzi, G. COVID-19 and Italy: What next? Lancet 2020, 395, 1225–1228.
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118.
- Hulme, K.D.; Gallo, L.A.; Short, K.R. Influenza Virus and Glycemic Variability in Diabetes: A Killer Combination? Front. Microbiol. 2017, 8, 861.
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139.
- Longato, E.; Di Camillo, B.; Sparacino, G.; Saccavini, C.; Avogaro, A.; Fadini, G.P. Diabetes diagnosis from administrative claims and estimation of the true prevalence of diabetes among 4.2 million individuals of the Veneto region (North East Italy). Nutr. Metab. Cardiovasc. Dis. 2020, 30, 84–91.
- Stegenga, M.E.; van der Crabben, S.N.; Blumer, R.M.; Levi, M.; Meijers, J.C.; Serlie, M.J.; Tanck, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 2008, 112, 82–89.
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440.
- Jafar, N.; Edriss, H.; Nugent, K. The Effect of Short-Term Hyperglycemia on the Innate Immune System. Am. J. Med. Sci. 2016, 351, 201–211.
- Yan, Y.; Yang, F.; Zhu, X.; Wang, M.; Sun, Z.; Zhao, T.; Yang, X.; Zou, Y. Analysis of clinical features and pulmonary CT features of coronavirus disease 2019 (COVID-19) patients with diabetes mellitus. Endokrynol. Pol. 2020, 71, 367–375.
- Popov, D.; Simionescu, M. Alterations of lung structure in experimental diabetes, and diabetes associated with hyperlipidaemia in hamsters. Eur. Respir. J. 1997, 10, 1850–1858.
- Exley, M.A.; Hand, L.; O’Shea, D.; Lynch, L. Interplay between the immune system and adipose tissue in obesity. J. Endocrinol. 2014, 223, R41–R48.
- Polito, R.; Nigro, E.; Messina, A.; Monaco, M.L.; Monda, V.; Scudiero, O.; Cibelli, G.; Valenzano, A.; Picciocchi, E.; Zammit, C.; et al. Adiponectin and Orexin-A as a Potential Immunity Link Between Adipose Tissue and Central Nervous System. Front. Physiol. 2018, 9, 982.
- Nigro, E.; Stiuso, P.; Matera, M.G.; Monaco, M.L.; Caraglia, M.; Maniscalco, M.; Perrotta, F.; Mazzarella, G.; Daniele, A.; Bianco, A. The anti-proliferative effects of adiponectin on human lung adenocarcinoma A549 cells and oxidative stress involvement. Pulm. Pharmacol. Ther. 2019, 55, 25–30.
- Pecoraro, A.; Nigro, E.; Polito, R.; Monaco, M.L.; Scudiero, O.; Mormile, I.; Cesoni Marcelli, A.; Capasso, M.; Habetswallner, F.; Genovese, A.; et al. Total and High Molecular Weight Adiponectin Expression Is Decreased in Patients with Common Variable Immunodeficiency: Correlation with Ig Replacement Therapy. Front. Immunol. 2017, 8, 895.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9.
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448.
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8.
- Gaddam, R.R.; Chambers, S.; Bhatia, M. ACE and ACE2 in inflammation: A tale of two enzymes. Inflamm. Allergy Drug Targets 2014, 13, 224–234.
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116.
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374.
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676.
- Jones, E.S.; Vinh, A.; McCarthy, C.A.; Gaspari, T.A.; Widdop, R.E. AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol. Ther. 2008, 120, 292–316.
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427.
- Xue, D.; Li, Y.; Jiang, Z.; Deng, G.; Li, M.; Liu, X.; Wang, Y. A ROS-dependent and Caspase-3-mediated apoptosis in sheep bronchial epithelial cells in response to Mycoplasma Ovipneumoniae infections. Vet. Immunol. Immunopathol. 2017, 187, 55–63.
- Zhang, X.; Wu, M.; Jiang, H.; Hao, J.; Zhang, Q.; Zhu, Q.; Saren, G.; Zhang, Y.; Meng, X.; Yue, X. Angiotensin II upregulates endothelial lipase expression via the NF-kappa B and MAPK signaling pathways. PLoS ONE 2014, 9, e107634.
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421.
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ. Res. 2005, 97, 946–953.
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610.
- Yang, G.; Tan, Z.; Zhou, L.; Yang, M.; Peng, L.; Liu, J.; Cai, J.; Yang, R.; Han, J.; Huang, Y.; et al. Effects of Angiotensin II Receptor Blockers and ACE (Angiotensin-Converting Enzyme) Inhibitors on Virus Infection, Inflammatory Status, and Clinical Outcomes in Patients With COVID-19 and Hypertension: A Single-Center Retrospective Study. Hypertension 2020, 76, 51–58.
- Zhong, J.C.; Ye, J.Y.; Jin, H.Y.; Yu, X.; Yu, H.M.; Zhu, D.L.; Gao, P.J.; Huang, D.Y.; Shuster, M.; Loibner, H.; et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul. Pept. 2011, 166, 90–97.
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21.
- Perrotta, F.; Matera, M.G.; Cazzola, M.; Bianco, A. Severe respiratory SARS-CoV2 infection: Does ACE2 receptor matter? Respir. Med. 2020, 168, 105996.
- Ali, R.M.; Al-Shorbagy, M.Y.; Helmy, M.W.; El-Abhar, H.S. Role of Wnt4/beta-catenin, Ang II/TGFbeta, ACE2, NF-kappaB, and IL-18 in attenuating renal ischemia/reperfusion-induced injury in rats treated with Vit D and pioglitazone. Eur. J. Pharmacol. 2018, 831, 68–76.
- Zhang, W.; Li, C.; Liu, B.; Wu, R.; Zou, N.; Xu, Y.Z.; Yang, Y.Y.; Zhang, F.; Zhou, H.M.; Wan, K.Q.; et al. Pioglitazone upregulates hepatic angiotensin converting enzyme 2 expression in rats with steatohepatitis. Ann. Hepatol. 2013, 12, 892–900.
- Darwish, I.; Mubareka, S.; Liles, W.C. Immunomodulatory therapy for severe influenza. Expert. Rev. Anti Infect. Ther. 2011, 9, 807–822.
- Drucker, D.J. Coronavirus Infections and Type 2 Diabetes-Shared Pathways with Therapeutic Implications. Endocr. Rev. 2020, 41, bnaa011.
- Sestan, M.; Marinovic, S.; Kavazovic, I.; Cekinovic, D.; Wueest, S.; Turk Wensveen, T.; Brizic, I.; Jonjic, S.; Konrad, D.; Wensveen, F.M.; et al. Virus-Induced Interferon-gamma Causes Insulin Resistance in Skeletal Muscle and Derails Glycemic Control in Obesity. Immunity 2018, 49, 164–177.e6.
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841.
- van Crevel, R.; van de Vijver, S.; Moore, D.A.J. The global diabetes epidemic: What does it mean for infectious diseases in tropical countries? Lancet Diabetes Endocrinol. 2017, 5, 457–468.
- Hodgson, K.; Morris, J.; Bridson, T.; Govan, B.; Rush, C.; Ketheesan, N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology 2015, 144, 171–185.
- Boccia, M.; Aronne, L.; Celia, B.; Mazzeo, G.; Ceparano, M.; D’Agnano, V.; Parrella, R.; Valente, T.; Bianco, A.; Perrotta, F. COVID-19 and coagulative axis: Review of emerging aspects in a novel disease. Monaldi Arch. Chest Dis. 2020, 90, 271–276.
- Shah, B.R.; Hux, J.E. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care 2003, 26, 510–513.
- Muniyappa, R.; Gubbi, S. COVID-19 pandemic, coronaviruses, and diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E736–E741.
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114.
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863.
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485.
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age 2009, 31, 285–291.
- Kim, S.J.; Mehta, H.H.; Wan, J.; Kuehnemann, C.; Chen, J.; Hu, J.F.; Hoffman, A.R.; Cohen, P. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging 2018, 10, 1239–1256.
- Gu, R.; Mao, T.; Lu, Q.; Tianjiao Su, T.; Wang, J. Myeloid dysregulation and therapeutic intervention in COVID-19. Semin. Immunol. 2021, 55, 101524.
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847.