Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Sabatino | -- | 2342 | 2024-02-21 10:37:12 | | | |
| 2 | Peter Tang | Meta information modification | 2342 | 2024-02-22 02:12:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sabatino, L.; Lapi, D.; Del Seppia, C. Thyroid Hormone Activity in the Brain. Encyclopedia. Available online: https://encyclopedia.pub/entry/55281 (accessed on 28 July 2026).
Sabatino L, Lapi D, Del Seppia C. Thyroid Hormone Activity in the Brain. Encyclopedia. Available at: https://encyclopedia.pub/entry/55281. Accessed July 28, 2026.
Sabatino, Laura, Dominga Lapi, Cristina Del Seppia. "Thyroid Hormone Activity in the Brain" Encyclopedia, https://encyclopedia.pub/entry/55281 (accessed July 28, 2026).
Sabatino, L., Lapi, D., & Del Seppia, C. (2024, February 21). Thyroid Hormone Activity in the Brain. In Encyclopedia. https://encyclopedia.pub/entry/55281
Sabatino, Laura, et al. "Thyroid Hormone Activity in the Brain." Encyclopedia. Web. 21 February, 2024.
Copy Citation
Thyroid hormones (THs) are essential in normal brain development, and cognitive and emotional functions. THs act through a cascade of events including uptake by the target cells by specific cell membrane transporters, activation or inactivation by deiodinase enzymes, and interaction with nuclear thyroid hormone receptors. Several thyroid responsive genes have been described in the developing and in the adult brain and many studies have demonstrated a systemic or local reduction in TH availability in neurologic disease and after brain injury.
thyroid hormones
TH deiodinases
TH transporters
brain damage
cognitive impairment
1. Introduction
THs regulate differentiation, growth, and energy metabolism in virtually all cells and tissues in all vertebrates, by affecting the expression of different sets of genes. In the brain, THs are essential for correcting brain maturation. They influence neurogenesis, neuronal and glial cell differentiation and migration, synaptogenesis, and myelination. Thyroid hormone deficiency may severely affect the brain during fetal and postnatal development, causing retarded maturation, intellectual deficits, and neurological impairment [1][2][3][4].
Thyroxine (T4) is considered a prohormone which is converted to the active form, triiodothyronine (T3). THs are synthesized in the thyroid in large part in the form of T4 (in a ratio of approximately 14:1 with respect to T3), are released in the circulation mostly bound to transport proteins, and reach the target tissues where specific transporters mediate the uptake of TH by the cells [5][6][7][8].
At the central level, TH homeostasis is regulated by the hypothalamic–pituitary–thyroid (HPT) axis through the activity of TSH-releasing hormone (TRH), Thyroid Stimulating Hormone (TSH), and the negative feedback on the HPT axis by circulating T3 and T4. Conversely, at the cellular level, T3 and T4 undergo to a strict homeostatic control relying on integrated transporters activity mediating the uptake of TH inside the cell, and on the three iodothyronine deiodinase enzymes, D1, D2, and D3, which regulate the activation and inactivation of TH and their metabolites [9][10] (Figure 1).
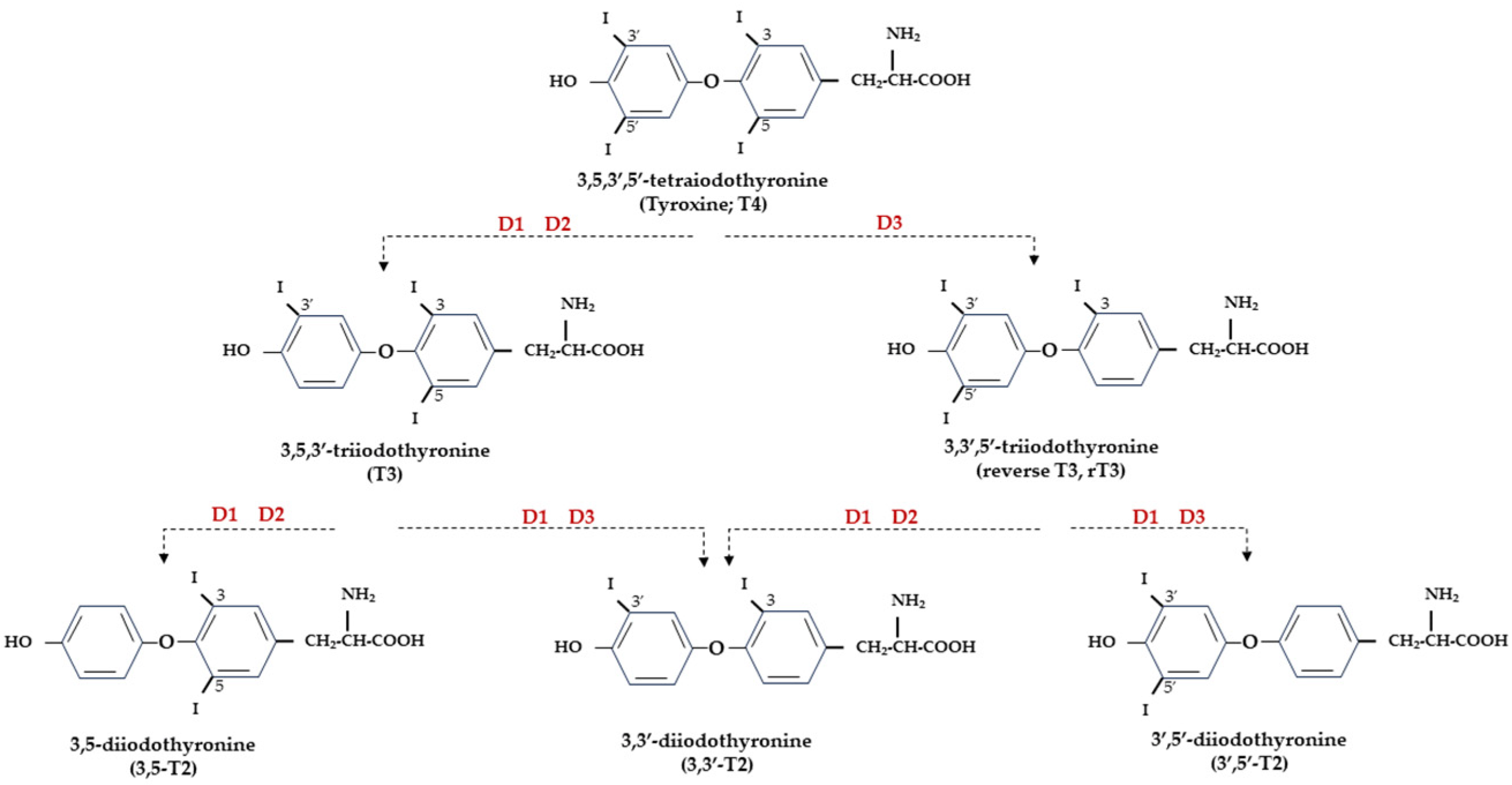
Figure 1. Representation of THs and derived iodothyronines. Deiodinases involved in each type of reaction are indicated as D1, D2, and D3.
2. THs and Normal Brain Development
THs are essential for normal brain development as they regulate the differentiation, migration, maturation, signaling, and metabolism of neurons. The function of THs is to provide the right temporal signal for the different stages of brain development to proceed, and the integration of different neuronal systems and association of glia cells to occur [11]. An incorrect synchronism in such developmental processes would inevitably lead to permanent deleterious outcomes. TH action is particularly critical starting from the late fetal stages until 3–4 weeks of the postnatal period in rodents [12], and the first postnatal month in humans [3]. For these reasons, most research on the role of THs in brain development has concentrated on the perinatal and early postnatal period and both the absence or the excess of THs in this critical time window can cause significant and irreversible structural and functional damage, with a relevant impact on the efficiency of the neurotransmitter system [13]. The presence of TH signaling components at very early stages of brain development, before the onset of fetal thyroid function, points to the maternal THs supply as a determining factor for proper fetal brain development in the early stages [14]. Interestingly, human data indicate that the maternal contribution to the fetus, even though in a smaller percentage, fulfils an important protective role until birth [3]. In fact, children born to mothers with thyroid disease experience an increased risk of neurologic and psychiatric diseases later in life [15][16]. Many of these problems are the expression of impaired TH signaling at different component levels, such as TH transporters, deiodinases, and TRs, but also reflect the alteration in the development of neurotransmitters in the central nervous system [17].
3. Nuclear TH Receptors
The genomic action of THs requires the mediation of nuclear TRs, which are ligand-activated transcription factors directly interacting with thyroid responding elements (TREs) present in the promoters of target genes and regulating their transcription [18]. Three main hormone-binding TRs are encoded by TRα and TRβ genes (THRA and THRB for human genes, Thra and Thrb for mice/rat genes) by alternative splicing (TRα1, TRβ1, and TRβ2) and show different affinities for the TRE sequences, but also for proteins and co-factors involved in gene transcription regulation [19]. The three main isoforms of TRs are expressed at variable levels in the brain. TRα1 is the predominant subtype; it is almost ubiquitous and is expressed since the earliest stages of development [20], while TRβ1 is the least expressed in the brain, and TRβ2, expressed in the hypothalamus, pituitary, cochlea, and retina, is considered responsible for T3-dependent negative feedback for TRH hypothalamic production and, consequently, for TSH release by the pituitary [21][22]. TRs account for many important functions of THs in embryonic and adult life and can interact with DNA both in the presence (activation) or absence (repression) of ligands [19][23]. Furthermore, non-T3 binding TRα isoforms have been described (TRα1 and TRα2) in the fetal, neonatal, and adult brain. Unliganded receptors (aporeceptors) are not silent but may have an opposite effect with respect to liganded receptors (holoreceptors); therefore, a T3-positively regulated target gene may be likely repressed by the aporeceptor and vice versa [23]. This is particularly relevant in the developing brain, where the control of T3 levels must be carefully calibrated and unliganded receptors may have a critical regulatory role in neuronal differentiation [24].
4. TH Transporters
Free THs enter the cells through transmembrane transporter proteins [25]. Many different carriers, belonging to several families, have been described to mediate TH transport across the plasma membrane [8][26]. Among them, the monocarboxylate transporter 8 (MCT8) is considered very specific for THs, with a higher affinity for T3 than T4 [27][28]; it is expressed in many tissues and organs and is determinant in the increase in TH intracellular availability [28][29]. Furthermore, MCT8 is also important for the transport of inactive metabolites such as reverse T3 (rT3) and 3,3′-diiodothyronine (T2) [28]. A mutation in the gene for MCT8 (SLC16A2) results in the impairment of T3 uptake in the neurons, leading to the neurological deficit known as Allan–Herndon–Dudley syndrome (AHDS), an X-linked disorder, characterized by hypotonia, spasticity, muscle weakness, neurological disorders, and cognitive impairment [30][31][32]. AHDS patients have high levels of T3, with borderline low T4 and high/normal TSH, with symptoms of hyperthyroidism in the peripheral tissues and of hypothyroidism at the central nervous system level. Moreover, the expression of MCT8 at the blood–brain barrier (BBB) level also suggests the role of this transporter in the T3 uptake by BBB [33]. Mouse models are commonly used to study intracellular TH signaling; however, although the expression pattern of MCT8 in the mice is similar to that of humans, Mct8-deficient mice do not show a neurological phenotype analogous to humans but only the same endocrine alterations observed in humans. Interestingly, in the brain of mice deprived of Mct8 function, the uptake of exogenous T3 is severely reduced, whereas T4 uptake is reduced by 50% with respect to the wild type [34]. These findings suggest that additional TH transporters, called “secondary TH transporters”, might be involved to compensate for Mct8 deprivation in mice [33][34]. Primary TH transporters are extremely specific for TH uptake by the target cells, whereas the secondary TH transporters can also mediate the uptake into the cells of various kinds of compounds, other than THs, and include the transporter of aromatic amino acids (MCT10), organic anion transporting polypeptides (OATPs), and the large neutral amino acid transporters (LAT1 and LAT2) [27][35][36]. OATP2B1, OATP3A1, and OATP4A1 are expressed ubiquitously, whereas other members of the OATP family, including OATP1B1, OATP1B3, and OATP1C1, have a more restricted expression, the latter being predominantly localized in the capillary endothelium and choroid plexus [37]. Furthermore, OATP1C1 is considered the principal responsible for T4 uptake from the circulation to the brain across the BBB, where it is converted to T3 and then transported into neurons by MCT8 [38][39]. At the brain level, LAT1 and LAT2 are expressed in the luminal and abluminal membranes of brain capillary endothelial cells of the BBB, and LAT1 is considered the most active isoform [40]. The system of TH transporters is very interesting from an evolutionary point of view since it shows how structurally different protein families can converge to the same common function (Figure 2).
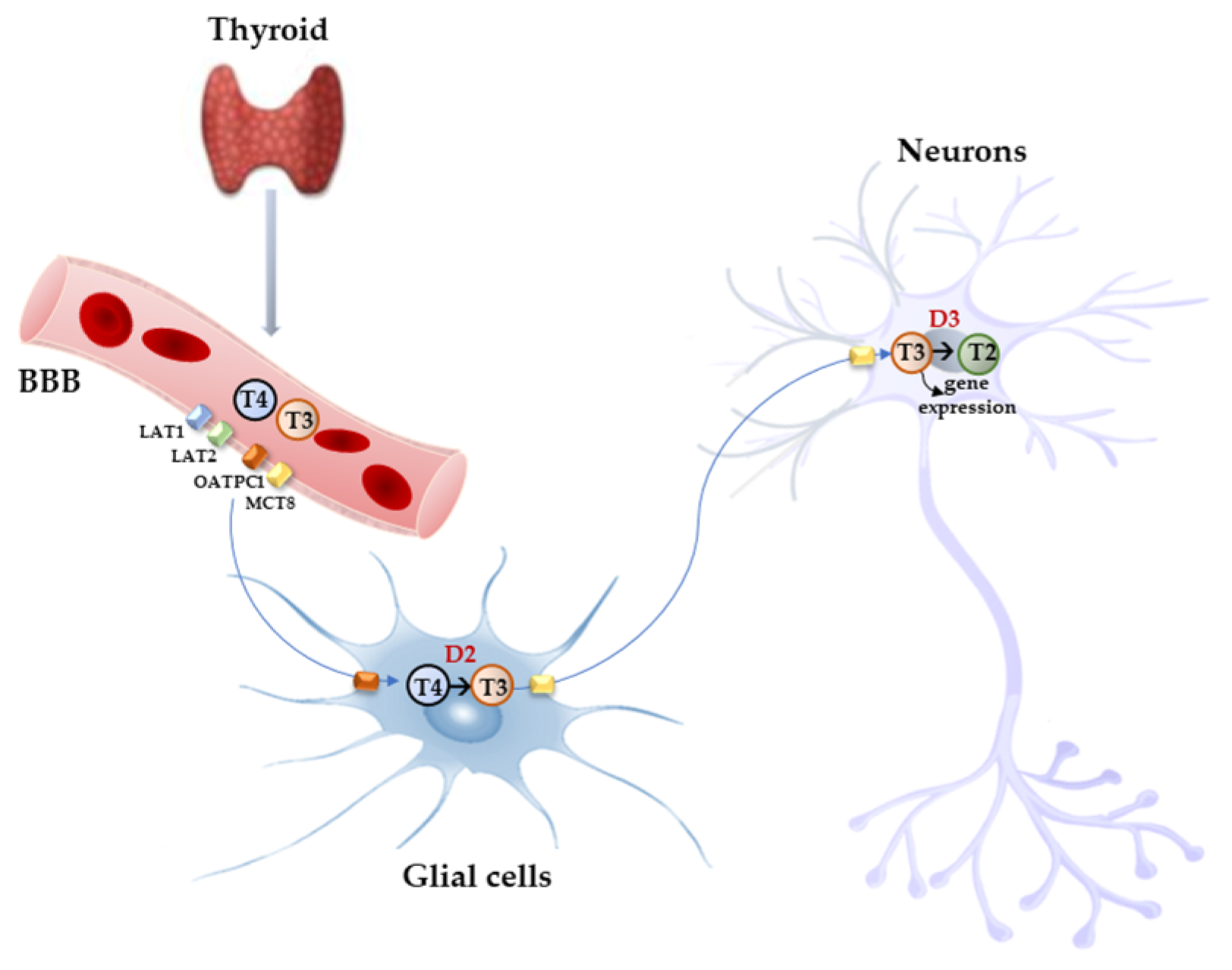
Figure 2. Model proposed for TH signaling in the brain. THs produced by the thyroid cross the BBB through TH transporters (OATP1C1, MCT8, LAT1, and LAT2) and enter astrocytes and tanycytes, where T4 is converted into active T3 by D2 and T3, in turn, with the mediation of MCT8 transporter reaches the adjacent neurons where it regulates the expression of target genes. In the neurons, T3 is deiodinated into 3,3′-T2 by D3 enzyme.
At the moment, no effective therapy is available to prevent or treat AHDS; however, several efforts have been made to find some TR-activating compounds which can cross the BBB and enter the target cells, independently from MCT8. An efficient treatment of this syndrome should be able to correct the hypothyroidism state in the central nervous system and the hypermetabolic condition due to an excess of T3 at the peripheral tissues. The first tested thyromimetic compound was 3,5-diiodothyropropionic acid (DITPA), which, once administered to Mct8-knockout (KO) mice, enters the liver and brain and is able to restore the correct TH signaling in the cells, reducing the excessive expression of T3-responsive genes and regulating D1 and D2 activities in a dose-dependent manner [41][42]. Other compounds studied in the context of AHDS are 3,3′,5-triiodothyroacetic acid (Triac), and 3,3′,5,5′-tetraiodothyroacetic acid (Tetrac), two naturally occurring metabolites of THs, which are less concentrated and with shorter half-lives than T4 and T3 [43]. As the other analogues, Triac is transported into the brain cells by a transporter different from MCT8 and is efficiently metabolized by D1 and D3 and affects the expression of TH-responsive genes in neurons [44]. However, Mct8-KO mice do not reproduce the neurological phenotype observed in AHDS patients, which makes this model not adequately reliable for what is observed in humans.
To overcome this experimental limit, an Mct8/Oatp1c1 double KO (DKO) mouse model was developed, in which the compensating mechanism of T4 to T3 conversion, mediated by D2 and observed in Mct8-KO mice, is also disrupted [45][46][47]. These DKO mice show both peripheral TH homeostasis and neurocognitive phenotype impairment and are therefore considered the most representative mouse model of AHDS [45][46]. In particular, DKO mice showed altered Purkinje cell dendritogenesis, with compromised cerebellar development and myelination, altered deiodinase activities, and TH target gene expression [45][46].
To date, complete phenotypical and neurological data on MCT8 deficiency are still lacking, due to the different approaches used in different studies; therefore, important pathophysiological aspects remain undefined and treatment options are limited. In a recent trial on pediatric and adult patients with MCT8 deficiency, some improving effects were reported after Triac treatment on peripheral thyrotoxicosis and neurocognitive outcomes [48]. Further studies on long-term Triac treatment in real-life settings confirmed the beneficial effects of Triac on the peripheral phenotype of MCT8-deficient patients and its clinical potential in AHDS [49].
5. TH Deiodinases
TH deiodinases are crucial in the functional diversification of TH signaling and are main actors in the fine regulation of TH homeostasis. The three deiodinases are synthesized by three different genes, which have high sequence similarity. The active site of the three enzymes is highly conserved and contains the rare selenocysteine amino acid, which is important for enzymatic activity. The three deiodinases are membrane homodimers, and the active site is oriented towards the cytosol [50]. More specifically, D1 and D3 are located in the plasma membrane, whereas D2 is in the endoplasmic reticulum membrane [51]. D1 and D2 both catalyse the 5′-phenolic ring deiodination of T4 and, thus, its conversion to T3. The two enzymes have different efficiency since D2 Km for T4 is in the nanomolar range, whereas D1 is in the micromolar range [5][52][53]. Furthermore, D1 has a longer half-life (12 h) than D2 (20–30 min) [54][55] and, in normal conditions, T4 is a better substrate for D2 than for D1. However, D1 (but not D2) has a dual specificity since it also allows the 5-tyrosyl ring deiodination of THs, thus acting as a scavenger enzyme in peripheral tissues to deiodinate iodothyronines (including sulphated iodothyronines) as well as other derivatives, clearing these compounds from circulation and recycling freed iodine [56]. About 20% of circulating T3 in humans (and approximately 50% in rats) is secreted by the thyroid, either through direct synthesis or by T4 to T3 deiodination by D1 and D2, while peripheral T4 to T3 conversion accounts for the remaining 80% [55]. The fact that in euthyroid patients the administration of propylthiouracil (PTU), a D1-selective inhibitor, reduces plasma levels of T3 by 20–30%, while in hyperthyroid patients the estimated reduction of plasma T3 is about 50%, supports the conclusion that D1 is the major responsible for circulating T3 increase in hyperthyroidism [57]. D2 is considered the deiodinase mainly responsible for local production of T3 in the tissues, and its activity increases in hypothyroid conditions, whereas in hyperthyroidism it is inactivated by selective ubiquination [58]. D1 and D2 are both important for TH homeostasis; however, data obtained from double Dio1 and Dio2 KO mice showed that these deiodinases were not essential for maintaining plasma T3 within the normal range, since compensatory mechanisms such as TSH-induced secretion of T3 by the thyroid and altered clearance of iodothyronines can be activated [56]. The brain relies on D2 activity for T3 availability; in fact, about 80% of brain T3 is produced locally in glial cells lining the third ventricle (astrocytes and tanycytes), where T4 is taken up after crossing the BBB and converted to the active hormone T3 that is distributed via Mct8 to the neighboring neurons where it exerts its regulatory action [59][60][61][62][63]. In tanycytes, more than astrocytes, D2 is believed to play an important role in T3 supply to nuclei of the hypothalamus and, thus, in feedback regulation on TRH-expressing neurons [64]. After selective inactivation of Dio2 in astrocytes (Astro-D2KO mouse model), in fact, the HPT axis is preserved due to D2 activity present in tanycytes [59]. Nonetheless, in Astro-D2KO mice, important alterations in T3-responsive genes were observed, leading to altered expression of specific gene sets in the hippocampus, and consequent mood and behavioural disorders [61].
References
- Porterfield, S.P.; Hendrich, C.E. The role of thyroid hormones in prenatal and neonatal neurological development—Current perspectives. Endocr. Rev. 1993, 14, 94–106.
- Oppenheimer, J.H.; Schwartz, H.L. Molecular basis of thyroid hormone-dependent brain development. Endocr. Rev. 1997, 18, 462–475.
- de Escobar, G.M.; Obregon, M.J.; del Rey, F.E. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract. Res. Clin. Endocrinol. Metabol. 2004, 18, 225–248.
- Jones, S.A.; Thoemke, K.R.; Anderson, G.W. The role of thyroid hormone in fetal and neonatal brain development. Curr. Opin. Endocrinol. Diabetes 2005, 12, 10–16.
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89.
- Wirth, E.K.; Schweizer, U.; Köhrle, J. Transport of thyroid hormone in brain. Front. Endocrinol. 2014, 5, 98.
- Köhrle, J. Thyroid hormone transporters in health and disease: Advances in thyroid hormone deiodination. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 173–191.
- Tedeschi, L.; Vassalle, C.; Iervasi, G.; Sabatino, L. Main Factors Involved in Thyroid Hormone Action. Molecules 2021, 26, 7337.
- Sabatino, L.; Vassalle, C.; Del Seppia, C.; Iervasi, G. Deiodinases and the Three Types of Thyroid Hormone Deiodination Reactions. Endocrinol. Metab. 2021, 36, 952–964.
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeold, A.; Bianco, A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 2008, 29, 898–938.
- Anderson, G.W.; Schoonover, C.M.; Jones, S.A. Control of thyroid hormone action in the developing rat brain. Thyroid 2003, 13, 1039–1056.
- Bernal, J. Thyroid hormones and brain development. Vitam. Horm. 2005, 71, 95–122.
- Alcaide, M.A.; Mayerl, S. Local thyroid hormone action in brain development. Int. J. Mol. Sci. 2023, 24, 12352.
- Miranda, A.; Sousa, N. Maternal hormonal milieu influence on fetal brain development. Brain Behav. 2018, 8, e00920.
- Andersen, S.L.; Laurberg, P.; Wu, C.S.; Olsen, J. Attention deficit hyperactivity disorder and autism spectrum disorder in children born to mothers with thyroid dysfunction: A Danish nationwide cohort study. BJOG 2014, 121, 1365–1374.
- Andersen, S.L.; Olsen, J.; Wu, C.S.; Laurberg, P. Psychiatric disease in late adolescence and young adulthood. Foetal programming by maternal hypothyroidism? Clin. Endocrinol. 2014, 81, 126–133.
- Powell, E.M. Interneuron development and epilepsy: Early genetic defects cause long-term consequences in seizures and susceptibility. Epilepsy Curr. 2013, 13, 172–176.
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170.
- Flamant, F.; Gauthier, K. Thyroid hormone receptors: The challenge of elucidating isotype-specific functions and cell-specific response. Biochim. Biophys. Acta 2013, 1830, 3900–3907.
- Wallis, K.; Dudazy, S.; van Hogerlinden, M.; Nordstrom, K.; Mittag, J.; Vennstrom, B. The thyroid hormone receptor 1 protein is expressed in embryonic postmitotic neurons and persists in most adult neurons. Mol. Endocrinol. 2010, 24, 1904–1916.
- Lazar, M.A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 1993, 14, 184–193.
- Liu, Y.Y.; Brent, G.A. The Role of Thyroid Hormone in Neuronal Protection. Compr. Physiol. 2021, 11, 2075–2095.
- Bernal, J.; Morte, B. Thyroid hormone receptor activity in the absence of ligand: Physiological and developmental implications. Biochim. Biophys. Acta 2013, 1830, 3893–3899.
- Muñoz, A.; Wrighton, C.; Seliger, B.; Bernal, J.; Beug, H. Thyroid hormone receptor/c-erbA: Control of commitment and differentiation in the neuronal/chromaffin progenitor line PC12. J. Cell Biol. 1993, 121, 423–438.
- Bernal, J.; Guadaño-Ferraz, A.; Morte, B. Thyroid hormone transport—Functions and clinical implications. Nat. Rev. Endocrinol. 2015, 11, 406–417.
- Braun, D.; Schweizer, U. Thyroid Hormone Transport and Transporters. In Vitamins and Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 106, pp. 19–44.
- Kinne, A.; Schülein, R.; Krause, G. Primary and secondary thyroid hormone transporters. Thyroid Res. 2011, 4, S7.
- Friesema, E.C.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 2003, 278, 40128–40135.
- Friesema, E.C.; Kuiper, G.G.; Jansen, J.; Visser, T.J.; Kester, M.H. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol. Endocrinol. 2006, 20, 2761–2772.
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Barrett, T.; Mancilla, E.E.; Svensson, J.; Kester, M.H.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437.
- Dumitrescu, A.M.; Liao, X.H.; Best, T.B.; Brockmann, K.; Refetoff, S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet. 2004, 74, 168–175.
- Schwartz, C.E.; Stevenson, R.E. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 307–321.
- Roberts, L.M.; Woodford, K.; Zhou, M.; Black, D.S.; Haggerty, J.E.; Tate, E.H.; Grindstaff, K.K.; Mengesha, W.; Raman, C.; Zerangue, N. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008, 149, 6251–6261.
- Wirth, E.K.; Roth, S.; Blechschmidt, C.; Holter, S.M.; Becker, L.; Racz, I.; Zimmer, A.; Klopstock, T.; Gailus-Durner, V.; Fuchs, H.; et al. Neuronal 3’,3,5- triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J. Neurosci. 2009, 29, 9439–9449.
- Friesema, E.C.; Jansen, J.; Jachtenberg, J.W.; Visser, W.E.; Kester, M.H.; Visser, T.J. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol. Endocrinol. 2008, 22, 1357–1369.
- Groeneweg, S.; van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, bnz008.
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287.
- Heuer, H.; Maier, M.K.; Iden, S.; Mittag, J.; Friesema, E.C.; Visser, T.J.; Bauer, K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 2005, 146, 1701–1706.
- Visser, W.E.; Friesema, E.C.; Jansen, J.; Visser, T.J. Thyroid hormone transport in and out of cells. Trends Endocrinol. Metab. 2008, 19, 50–56.
- Killian, D.M.; Chikhale, P.J. Predominant functional activity of the large, neutral amino acid transporter LAT1 isoform at the cerebrovasculature. Neurosci. Lett. 2001, 306, 1–4.
- Di Cosmo, C.; Liao, X.H.; Dumitrescu, A.M.; Weiss, R.E.; Refetoff, S. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology 2009, 150, 4450–4458.
- Ferrara, A.M.; Liao, X.H.; Ye, H.; Weiss, R.E.; Dumitrescu, A.M.; Refetoff, S. The Thyroid Hormone Analog DITPA Ameliorates Metabolic Parameters of Male Mice with Mct8 Deficiency. Endocrinology 2015, 156, 3889–3894.
- Groeneweg, S.; Peeters, R.P.; Visser, T.J.; Visser, W.E. Therapeutic applications of thyroid hormone analogues in resistance to thyroid hormone (RTH) syndromes. Mol. Cell. Endocrinol. 2017, 458, 82–90.
- Kersseboom, S.; Horn, S.; Visser, W.E.; Chen, J.; Friesema, E.C.; Vaurs-Barrière, C.; Peeters, R.P.; Heuer, H.; Visser, T.J. In Vitro and Mouse Studies Supporting Therapeutic Utility of Triiodothyroacetic Acid in MCT8 Deficiency. Mol. Endocrinol. 2014, 28, 1961–1970.
- van Geest, F.S.; Gunhanlar, N.; Groeneweg, S.; Visser, W.E. Monocarboxylate Transporter 8 Deficiency: From Pathophysiological Understanding to Therapy Development. Front. Endocrinol. (Lausanne) 2021, 12, 723750.
- Mayerl, S.; Müller, J.; Bauer, R.; Richert, S.; Kassmann, C.M.; Darras, V.M.; Buder, K.; Boelen, A.; Visser, T.J.; Heuer, H. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Investig. 2014, 124, 1987–1999.
- Morte, B.; Gil-Ibañez, P.; Heuer, H.; Bernal, J. Brain Gene Expression in Systemic Hypothyroidism and Mouse Models of MCT8 Deficiency: The Mct8-Oatp1c1-Dio2 Triad. Thyroid 2021, 31, 985–993.
- Groeneweg, S.; Peeters, R.P.; Moran, C.; Stoupa, A.; Auriol, F.; Tonduti, D.; Dica, A.; Paone, L.; Rozenkova, K.; Malikova, J.; et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 695–706.
- van Geest, F.S.; Groeneweg, S.; van den Akker, E.L.T.; Bacos, I.; Barca, D.; van den Berg, S.A.A.; Bertini, E.; Brunner, D.; Brunetti-Pierri, N.; Cappa, M.; et al. Long-Term Efficacy of T3 Analogue Triac in Children and Adults with MCT8 Deficiency: A Real-Life Retrospective Cohort Study. J. Clin. Endocrinol. Metab. 2022, 107, e1136–e1147.
- Köhrle, J.; Frädrich, C. Deiodinases control local cellular and systemic thyroid hormone availability. Free Radic. Biol. Med. 2022, 193, 59–79.
- Gereben, B.; Zeold, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell. Mol. Life Sci. 2008, 65, 570–590.
- Sabatino, L.; Iervasi, G.; Ferrazzi, P.; Francesconi, D.; Chopra, I.J. A study of iodothyronine 5′-monodeiodinase activities in normal and pathological tissues in man and their comparison with activities in rat tissues. Life Sci. 2000, 68, 191–202.
- Sabatino, L.; Chopra, I.J.; Tanavoli, S.; Iacconi, P.; Iervasi, G. A radioimmunoassay for type I iodothyronine 5′-monodeiodinase in human tissues. Thyroid 2001, 11, 733–739.
- Maia, A.L.; Kim, B.W.; Huang, S.A.; Harney, J.W.; Larsen, P.R. Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J. Clin. Investig. 2005, 115, 2524–2533.
- Galton, V.A.; Schneider, M.J.; Clark, A.S.; St Germain, D.L. Life without thyroxine to 3,5,3ʹ- triiodothyronine conversion: Studies in mice devoid of the 5ʹ-deiodinases. Endocrinology 2009, 150, 2957–2963.
- Pilo, A.; Iervasi, G.; Vitek, F.; Ferdeghini, M.; Cazzuola, F.; Bianchi, R. Thyroidal and peripheral production of 3,5,3′-triiodothyronine in humans by multicompartmental analysis. Am. J. Physiol. 1990, 258, E715–E726.
- Laurberg, P.; Tørring, J.; Weeke, J. A comparison of the effects of propylthiouracil and methimazol on circulating thyroid hormones and various measures of peripheral thyroid hormone effects in thyrotoxic patients. Acta Endocrinol. (Copenh) 1985, 108, 51–54.
- Gereben, B.; Goncalves, C.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. Selective proteolysis of human type 2 deiodinase: A novel ubiquitin-proteasomal mediated mechanism for regulation of hormone activation. Mol. Endocrinol. 2000, 14, 1697–1708.
- Sánchez, E.; Vargas, M.A.; Singru, P.S.; Pascual, I.; Romero, F.; Fekete, C.; Charli, J.L.; Lechan, R.M. Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology 2009, 150, 2283–2291.
- Freitas, B.C.; Gereben, B.; Castillo, M.; Kalló, I.; Zeöld, A.; Egri, P.; Liposits, Z.; Zavacki, A.M.; Maciel, R.M.; Jo, S.; et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J. Clin. Investig. 2010, 120, 2206–2217.
- Bocco, B.M.; Werneck-de-Castro, J.P.; Oliveira, K.C.; Fernandes, G.W.; Fonseca, T.L.; Nascimento, B.P.; McAninch, E.A.; Ricci, E.; Kvárta-Papp, Z.; Fekete, C.; et al. Type 2 Deiodinase Disruption in Astrocytes Results in Anxiety-Depressive-Like Behavior in Male Mice. Endocrinology 2016, 157, 3682–3695.
- Fonseca, T.L.; Correa-Medina, M.; Campos, M.P.; Wittmann, G.; Werneck-de-Castro, J.P.; Arrojo e Drigo, R.; Mora-Garzon, M.; Ueta, C.B.; Caicedo, A.; Fekete, C.; et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J. Clin. Investig. 2013, 123, 1492–1500.
- Guadaño-Ferraz, A.; Obregon, M.J.; Germain, D.L.S.; Bernal, J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc. Natl. Acad. Sci. USA 1997, 94, 10391–10396.
- Rodriguez-Rodriguez, A.; Lazcano, I.; Sanchez-Jaramillo, E.; Uribe, R.M.; Jaimes-Hoy, L.; Joseph-Bravo, P.; Charli, J.L. Tanycytes and the Control of Thyrotropin-Releasing Hormone Flux into Portal Capillaries. Front. Endocrinol. 2019, 10, 401.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
639
Revisions:
2 times
(View History)
Update Date:
22 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No