Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Laura Sabatino and Version 2 by Peter Tang.
Thyroid hormones (THs) are essential in normal brain development, and cognitive and emotional functions. THs act through a cascade of events including uptake by the target cells by specific cell membrane transporters, activation or inactivation by deiodinase enzymes, and interaction with nuclear thyroid hormone receptors. Several thyroid responsive genes have been described in the developing and in the adult brain and many studies have demonstrated a systemic or local reduction in TH availability in neurologic disease and after brain injury.
- thyroid hormones
- TH deiodinases
- TH transporters
- brain damage
- cognitive impairment
1. Introduction
THs regulate differentiation, growth, and energy metabolism in virtually all cells and tissues in all vertebrates, by affecting the expression of different sets of genes. In the brain, THs are essential for correcting brain maturation. They influence neurogenesis, neuronal and glial cell differentiation and migration, synaptogenesis, and myelination. Thyroid hormone deficiency may severely affect the brain during fetal and postnatal development, causing retarded maturation, intellectual deficits, and neurological impairment [1][2][3][4][1,2,3,4].
Thyroxine (T4) is considered a prohormone which is converted to the active form, triiodothyronine (T3). THs are synthesized in the thyroid in large part in the form of T4 (in a ratio of approximately 14:1 with respect to T3), are released in the circulation mostly bound to transport proteins, and reach the target tissues where specific transporters mediate the uptake of TH by the cells [5][6][7][8][5,6,7,8].
At the central level, TH homeostasis is regulated by the hypothalamic–pituitary–thyroid (HPT) axis through the activity of TSH-releasing hormone (TRH), Thyroid Stimulating Hormone (TSH), and the negative feedback on the HPT axis by circulating T3 and T4. Conversely, at the cellular level, T3 and T4 undergo to a strict homeostatic control relying on integrated transporters activity mediating the uptake of TH inside the cell, and on the three iodothyronine deiodinase enzymes, D1, D2, and D3, which regulate the activation and inactivation of TH and their metabolites [9][10][9,10] (Figure 1).
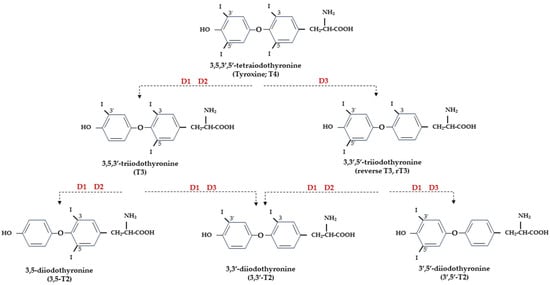
Figure 1.
Representation of THs and derived iodothyronines. Deiodinases involved in each type of reaction are indicated as D1, D2, and D3.
2. THs and Normal Brain Development
THs are essential for normal brain development as they regulate the differentiation, migration, maturation, signaling, and metabolism of neurons. The function of THs is to provide the right temporal signal for the different stages of brain development to proceed, and the integration of different neuronal systems and association of glia cells to occur [11][15]. An incorrect synchronism in such developmental processes would inevitably lead to permanent deleterious outcomes. TH action is particularly critical starting from the late fetal stages until 3–4 weeks of the postnatal period in rodents [12][16], and the first postnatal month in humans [3]. For these reasons, most research on the role of THs in brain development has concentrated on the perinatal and early postnatal period and both the absence or the excess of THs in this critical time window can cause significant and irreversible structural and functional damage, with a relevant impact on the efficiency of the neurotransmitter system [13][17]. The presence of TH signaling components at very early stages of brain development, before the onset of fetal thyroid function, points to the maternal THs supply as a determining factor for proper fetal brain development in the early stages [14][18]. Interestingly, human data indicate that the maternal contribution to the fetus, even though in a smaller percentage, fulfils an important protective role until birth [3]. In fact, children born to mothers with thyroid disease experience an increased risk of neurologic and psychiatric diseases later in life [15][16][19,20]. Many of these problems are the expression of impaired TH signaling at different component levels, such as TH transporters, deiodinases, and TRs, but also reflect the alteration in the development of neurotransmitters in the central nervous system [17][21].3. Nuclear TH Receptors
The genomic action of THs requires the mediation of nuclear TRs, which are ligand-activated transcription factors directly interacting with thyroid responding elements (TREs) present in the promoters of target genes and regulating their transcription [18][11]. Three main hormone-binding TRs are encoded by TRα and TRβ genes (THRA and THRB for human genes, Thra and Thrb for mice/rat genes) by alternative splicing (TRα1, TRβ1, and TRβ2) and show different affinities for the TRE sequences, but also for proteins and co-factors involved in gene transcription regulation [19][22]. The three main isoforms of TRs are expressed at variable levels in the brain. TRα1 is the predominant subtype; it is almost ubiquitous and is expressed since the earliest stages of development [20][23], while TRβ1 is the least expressed in the brain, and TRβ2, expressed in the hypothalamus, pituitary, cochlea, and retina, is considered responsible for T3-dependent negative feedback for TRH hypothalamic production and, consequently, for TSH release by the pituitary [21][22][24,25]. TRs account for many important functions of THs in embryonic and adult life and can interact with DNA both in the presence (activation) or absence (repression) of ligands [19][23][22,26]. Furthermore, non-T3 binding TRα isoforms have been described (TRα1 and TRα2) in the fetal, neonatal, and adult brain. Unliganded receptors (aporeceptors) are not silent but may have an opposite effect with respect to liganded receptors (holoreceptors); therefore, a T3-positively regulated target gene may be likely repressed by the aporeceptor and vice versa [23][26]. This is particularly relevant in the developing brain, where the control of T3 levels must be carefully calibrated and unliganded receptors may have a critical regulatory role in neuronal differentiation [24][27].4. TH Transporters
Free THs enter the cells through transmembrane transporter proteins [25][28]. Many different carriers, belonging to several families, have been described to mediate TH transport across the plasma membrane [8][26][8,29]. Among them, the monocarboxylate transporter 8 (MCT8) is considered very specific for THs, with a higher affinity for T3 than T4 [27][28][30,31]; it is expressed in many tissues and organs and is determinant in the increase in TH intracellular availability [28][29][31,32]. Furthermore, MCT8 is also important for the transport of inactive metabolites such as reverse T3 (rT3) and 3,3′-diiodothyronine (T2) [28][31]. A mutation in the gene for MCT8 (SLC16A2) results in the impairment of T3 uptake in the neurons, leading to the neurological deficit known as Allan–Herndon–Dudley syndrome (AHDS), an X-linked disorder, characterized by hypotonia, spasticity, muscle weakness, neurological disorders, and cognitive impairment [30][31][32][33,34,35]. AHDS patients have high levels of T3, with borderline low T4 and high/normal TSH, with symptoms of hyperthyroidism in the peripheral tissues and of hypothyroidism at the central nervous system level. Moreover, the expression of MCT8 at the blood–brain barrier (BBB) level also suggests the role of this transporter in the T3 uptake by BBB [33][36]. Mouse models are commonly used to study intracellular TH signaling; however, although the expression pattern of MCT8 in the mice is similar to that of humans, Mct8-deficient mice do not show a neurological phenotype analogous to humans but only the same endocrine alterations observed in humans. Interestingly, in the brain of mice deprived of Mct8 function, the uptake of exogenous T3 is severely reduced, whereas T4 uptake is reduced by 50% with respect to the wild type [34][37]. These findings suggest that additional TH transporters, called “secondary TH transporters”, might be involved to compensate for Mct8 deprivation in mice [33][34][36,37]. Primary TH transporters are extremely specific for TH uptake by the target cells, whereas the secondary TH transporters can also mediate the uptake into the cells of various kinds of compounds, other than THs, and include the transporter of aromatic amino acids (MCT10), organic anion transporting polypeptides (OATPs), and the large neutral amino acid transporters (LAT1 and LAT2) [27][35][36][30,38,39]. OATP2B1, OATP3A1, and OATP4A1 are expressed ubiquitously, whereas other members of the OATP family, including OATP1B1, OATP1B3, and OATP1C1, have a more restricted expression, the latter being predominantly localized in the capillary endothelium and choroid plexus [37][40]. Furthermore, OATP1C1 is considered the principal responsible for T4 uptake from the circulation to the brain across the BBB, where it is converted to T3 and then transported into neurons by MCT8 [38][39][41,42]. At the brain level, LAT1 and LAT2 are expressed in the luminal and abluminal membranes of brain capillary endothelial cells of the BBB, and LAT1 is considered the most active isoform [40][43]. The system of TH transporters is very interesting from an evolutionary point of view since it shows how structurally different protein families can converge to the same common function (Figure 2).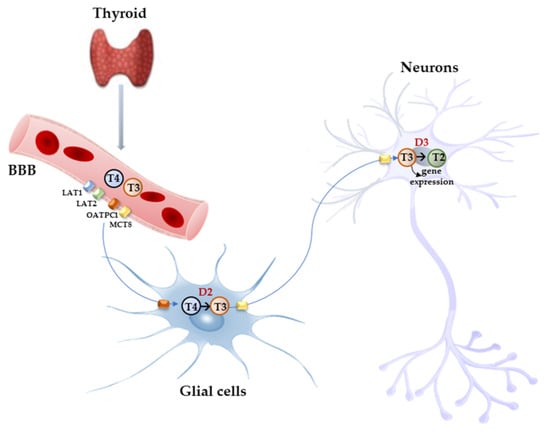
Figure 2. Model proposed for TH signaling in the brain. THs produced by the thyroid cross the BBB through TH transporters (OATP1C1, MCT8, LAT1, and LAT2) and enter astrocytes and tanycytes, where T4 is converted into active T3 by D2 and T3, in turn, with the mediation of MCT8 transporter reaches the adjacent neurons where it regulates the expression of target genes. In the neurons, T3 is deiodinated into 3,3′-T2 by D3 enzyme.