 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gabriella Nicolini | -- | 2676 | 2024-02-21 10:24:27 | | | |
| 2 | Lindsay Dong | Meta information modification | 2676 | 2024-02-23 04:05:36 | | |
Video Upload Options
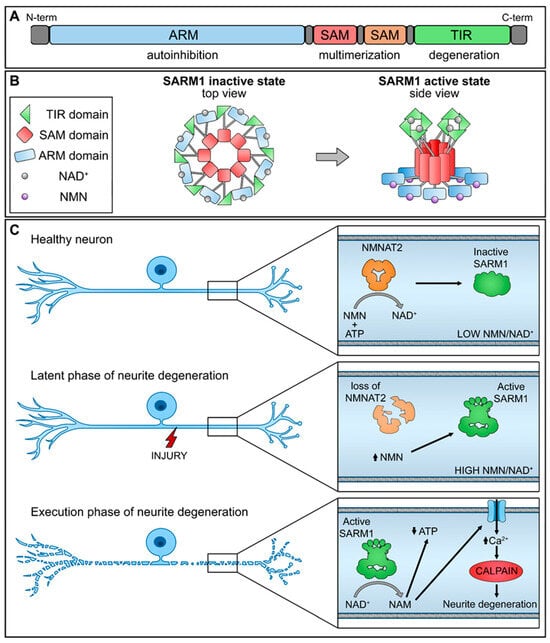
Chemotherapy-induced peripheral neuropathy (CIPN) commonly arises as a side effect of diverse cancer chemotherapy treatments. This condition presents symptoms such as numbness, tingling, and altered sensation in patients, often accompanied by neuropathic pain. Pathologically, CIPN is characterized by an intensive “dying-back” axonopathy, starting at the intra-epidermal sensory innervations and advancing retrogradely. The lack of comprehensive understanding regarding its underlying mechanisms explains the absence of effective treatments for CIPN. Recent investigations into axon degeneration mechanisms have pinpointed nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and sterile alpha and TIR motif-containing 1 protein (SARM1) as pivotal mediators of injury-induced axonal degeneration.
1. Introduction
2. NMNAT2/SARM1 and NAD+ Synthesis
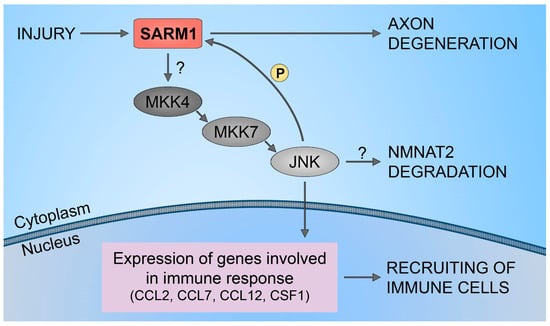
3. NMNAT2/SARM1 Downstream Pathways Regulation
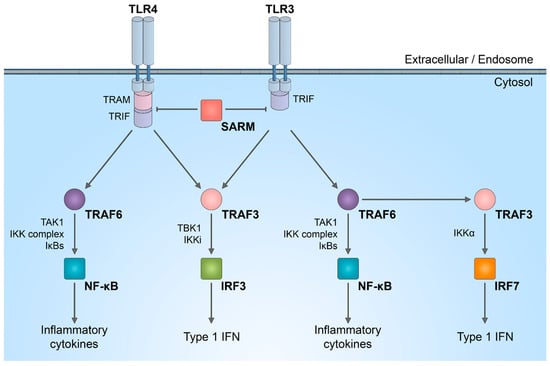
4. NMNAT2/SARM1 Role in Immune System Stimulation
5. NMNAT2/SARM1 in Chemotherapy-Induced Neuropathy
6. SARM1 Inhibitors
References
- Fortunato, C.; Mazzola, F.; Raffaelli, N. The key role of the NAD biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase in regulating cell functions. IUBMB Life 2022, 74, 562–572.
- Jayaram, H.N.; Kusumanchi, P.; Yalowitz, J.A. NMNAT expression and its relation to NAD metabolism. Curr. Med. Chem. 2011, 18, 1962–1972.
- Brunetti, L.; Di Stefano, M.; Ruggieri, S.; Cimadamore, F.; Magni, G. Homology modeling and deletion mutants of human nicotinamide mononucleotide adenylyltransferase isozyme 2: New insights on structure and function relationship. Protein Sci. 2010, 19, 2440–2450.
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341.
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLoS Biol. 2016, 14, e1002472.
- Li, W.; Gao, M.; Hu, C.; Chen, X.; Zhou, Y. NMNAT2: An important metabolic enzyme affecting the disease progression. Biomed. Pharmacother. 2023, 158, 114143.
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD. Mol. Metab. 2021, 49, 101195.
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622.
- Qi, J.; Cui, C.; Deng, Q.; Wang, L.; Chen, R.; Zhai, D.; Xie, L.; Yu, J. Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncol. Lett. 2018, 16, 5829–5837.
- Challa, S.; Khulpateea, B.R.; Nandu, T.; Camacho, C.V.; Ryu, K.W.; Chen, H.; Peng, Y.; Lea, J.S.; Kraus, W.L. Ribosome ADP-ribosylation inhibits translation and maintains proteostasis in cancers. Cell 2021, 184, 4531–4546.e26.
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 2016, 89, 449–460.
- Bradshaw, D.V.; Knutsen, A.K.; Korotcov, A.; Sullivan, G.M.; Radomski, K.L.; Dardzinski, B.J.; Zi, X.; McDaniel, D.P.; Armstrong, R.C. Genetic inactivation of SARM1 axon degeneration pathway improves outcome trajectory after experimental traumatic brain injury based on pathological, radiological, and functional measures. Acta Neuropathol. Commun. 2021, 9, 89.
- Marion, C.M.; McDaniel, D.P.; Armstrong, R.C. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol. 2019, 321, 113040.
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108.
- Gilley, J.; Orsomando, G.; Nascimento-Ferreira, I.; Coleman, M.P. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015, 10, 1974–1981.
- Carty, M.; Bowie, A.G. SARM: From immune regulator to cell executioner. Biochem. Pharmacol. 2019, 161, 52–62.
- Bratkowski, M.; Xie, T.; Thayer, D.A.; Lad, S.; Mathur, P.; Yang, Y.S.; Danko, G.; Burdett, T.C.; Danao, J.; Cantor, A.; et al. Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep. 2020, 32, 107999.
- Figley, M.D.; Gu, W.; Nanson, J.D.; Shi, Y.; Sasaki, Y.; Cunnea, K.; Malde, A.K.; Jia, X.; Luo, Z.; Saikot, F.K.; et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD. Neuron 2021, 109, 1118–1136.e11.
- Waller, T.J.; Collins, C.A. An NAD+/NMN balancing act by SARM1 and NMNAT2 controls axonal degeneration. Neuron 2021, 109, 1067–1069.
- Yang, J.; Wu, Z.; Renier, N.; Simon, D.J.; Uryu, K.; Park, D.S.; Greer, P.A.; Tournier, C.; Davis, R.J.; Tessier-Lavigne, M. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 2015, 160, 161–176.
- Hsu, J.M.; Kang, Y.; Corty, M.M.; Mathieson, D.; Peters, O.M.; Freeman, M.R. Injury-Induced Inhibition of Bystander Neurons Requires dSarm and Signaling from Glia. Neuron 2021, 109, 473–487.e5.
- Walker, L.J.; Summers, D.W.; Sasaki, Y.; Brace, E.J.; Milbrandt, J.; DiAntonio, A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. eLife 2017, 6, e22540.
- Asghari Adib, E.; Smithson, L.J.; Collins, C.A. An axonal stress response pathway: Degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol. 2018, 53, 110–119.
- Jin, Y.; Zheng, B. Multitasking: Dual Leucine Zipper-Bearing Kinases in Neuronal Development and Stress Management. Annu. Rev. Cell Dev. Biol. 2019, 35, 501–521.
- Cavalli, V.; Kujala, P.; Klumperman, J.; Goldstein, L.S. Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 2005, 168, 775–787.
- Lindwall, C.; Kanje, M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell Neurosci. 2005, 29, 269–282.
- Li, Y.; Pazyra-Murphy, M.F.; Avizonis, D.; de Sá Tavares Russo, M.; Tang, S.; Chen, C.Y.; Hsueh, Y.P.; Bergholz, J.S.; Jiang, T.; Zhao, J.J.; et al. Sarm1 activation produces cADPR to increase intra-axonal Ca++ and promote axon degeneration in PIPN. J. Cell Biol. 2022, 221, e202106080.
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; Hoving, J.J.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139.
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368.
- Liu, H.Y.; Chen, C.Y.; Hsueh, Y.P. Innate immune responses regulate morphogenesis and degeneration: Roles of Toll-like receptors and Sarm1 in neurons. Neurosci. Bull. 2014, 30, 645–654.
- Mukherjee, P.; Winkler, C.W.; Taylor, K.G.; Woods, T.A.; Nair, V.; Khan, B.A.; Peterson, K.E. SARM1, Not MyD88, Mediates TLR7/TLR9-Induced Apoptosis in Neurons. J. Immunol. 2015, 195, 4913–4921.
- Loring, H.S.; Thompson, P.R. Emergence of SARM1 as a Potential Therapeutic Target for Wallerian-type Diseases. Cell Chem. Biol. 2020, 27, 1–13.
- Tan, R.S.; Ho, B.; Leung, B.P.; Ding, J.L. TLR cross-talk confers specificity to innate immunity. Int. Rev. Immunol. 2014, 33, 443–453.
- Carty, M.; Goodbody, R.; Schröder, M.; Stack, J.; Moynagh, P.N.; Bowie, A.G. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 2006, 7, 1074–1081.
- Geisler, S.; Doan, R.A.; Cheng, G.C.; Cetinkaya-Fisgin, A.; Huang, S.X.; Höke, A.; Milbrandt, J.; DiAntonio, A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 2019, 4, e129920.
- Gould, S.A.; White, M.; Wilbrey, A.L.; Pór, E.; Coleman, M.P.; Adalbert, R. Protection against oxaliplatin-induced mechanical and thermal hypersensitivity in Sarm1. Exp. Neurol. 2021, 338, 113607.
- Cetinkaya-Fisgin, A.; Luan, X.; Reed, N.; Jeong, Y.E.; Oh, B.C.; Hoke, A. Cisplatin induced neurotoxicity is mediated by Sarm1 and calpain activation. Sci. Rep. 2020, 10, 21889.
- Turkiew, E.; Falconer, D.; Reed, N.; Höke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171.
- Loring, H.S.; Parelkar, S.S.; Mondal, S.; Thompson, P.R. Identification of the first noncompetitive SARM1 inhibitors. Bioorg. Med. Chem. 2020, 28, 115644.
- Khazma, T.; Golan-Vaishenker, Y.; Guez-Haddad, J.; Grossman, A.; Sain, R.; Weitman, M.; Plotnikov, A.; Zalk, R.; Yaron, A.; Hons, M.; et al. A duplex structure of SARM1 octamers stabilized by a new inhibitor. Cell Mol. Life Sci. 2022, 80, 16.
- Bosanac, T.; Hughes, R.O.; Engber, T.; Devraj, R.; Brearley, A.; Danker, K.; Young, K.; Kopatz, J.; Hermann, M.; Berthemy, A.; et al. Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 2021, 144, 3226–3238.
- Li, W.H.; Huang, K.; Cai, Y.; Wang, Q.W.; Zhu, W.J.; Hou, Y.N.; Wang, S.; Cao, S.; Zhao, Z.Y.; Xie, X.J.; et al. Permeant fluorescent probes visualize the activation of SARM1 and uncover an anti-neurodegenerative drug candidate. eLife 2021, 10, e67381.
- Feldman, H.C.; Merlini, E.; Guijas, C.; DeMeester, K.E.; Njomen, E.; Kozina, E.M.; Yokoyama, M.; Vinogradova, E.; Reardon, H.T.; Melillo, B.; et al. Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc. Natl. Acad. Sci. USA 2022, 119, e2208457119.

