Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aviv Philip Goncharov | -- | 3773 | 2024-02-20 19:28:12 | | | |
| 2 | Sirius Huang | -6 word(s) | 3767 | 2024-02-21 02:38:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Goncharov, A.P.; Vashakidze, N.; Kharaishvili, G. Epithelial-Mesenchymal Transition. Encyclopedia. Available online: https://encyclopedia.pub/entry/55248 (accessed on 03 July 2026).
Goncharov AP, Vashakidze N, Kharaishvili G. Epithelial-Mesenchymal Transition. Encyclopedia. Available at: https://encyclopedia.pub/entry/55248. Accessed July 03, 2026.
Goncharov, Aviv Philip, Nino Vashakidze, Gvantsa Kharaishvili. "Epithelial-Mesenchymal Transition" Encyclopedia, https://encyclopedia.pub/entry/55248 (accessed July 03, 2026).
Goncharov, A.P., Vashakidze, N., & Kharaishvili, G. (2024, February 20). Epithelial-Mesenchymal Transition. In Encyclopedia. https://encyclopedia.pub/entry/55248
Goncharov, Aviv Philip, et al. "Epithelial-Mesenchymal Transition." Encyclopedia. Web. 20 February, 2024.
Copy Citation
Epithelial-mesenchymal transition (EMT) is a crucial and fundamental mechanism in many cellular processes, beginning with embryogenesis via tissue remodulation and wound healing, and plays a vital role in tumorigenesis and metastasis formation. EMT is a complex process that involves many transcription factors and genes that enable the tumor cell to leave the primary location, invade the basement membrane, and send metastasis to other tissues. Moreover, it may help the tumor avoid the immune system and establish radioresistance and chemoresistance. It may also change the normal microenvironment, thus promoting other key factors for tumor survival, such as hypoxia-induced factor-1 (HIF-1) and promoting neoangiogenesis.
epithelial-mesenchymal transition
EMT
prostate cancer
BPH
transcription factors
1. Description of Types of EMT
Epithelial-mesenchymal transition (EMT) is a complex manifestation of epithelial plasticity [1]. EMT is a process that allows epithelial cells, which are typically attached to the basement membrane, to pass through various changes that enable them to acquire a mesenchymal cell phenotype. The process includes changes in cytoskeleton and cell shape, enhancing migration and establishment of metastatic potential, invasiveness, increased resistance to apoptosis, and significantly elevated production of extracellular matrix (ECM) components [2].
EMT was recognized in the late 70s and received distinct attention due to its physiological presence, such as embryonic development and pathological conditions. Over the years, a significant focus has been on the relationship between EMT and various cancers.
The current understanding of the signaling processes that mediate EMT is starting to provide new opportunities for utilizing the underlying molecular mechanisms to create novel treatments.
On the other hand, the reverse process, mesenchymal to epithelial transition (MET), also occurs and is essential in illustrating the potential of EMT to be a reversible process. Also, endothelial cells show properties similar to epithelial cells and can lose endothelial characteristics while gaining mesenchymal characteristics.
EMTs are related to three physiological and pathological scenarios with different consequences.
1.1. Type 1 EMT
Type 1 EMT is associated with embryogenesis and occurs at a few locations and stages in the process of organ development, e.g., during the gastrulation when ectodermal cells give rise to mesoderm and also during neural crest migration. Type 1 EMT generates new tissue with mesenchymal phenotype but was not found to cause other manifestations such as fibrosis or systemic invasion by high-grade tumors [3][4].
The coordination of EMT is enabled by various proteins such as Snail, Eomesodermin, and Mesoderm posterior protein. The Snail suppresses the E-cadherin, thus promoting the transition. This explains the origin of migratory neural crest cells from the neuroectodermal epithelial cells [5].
Developmental EMT is regulated by morphogenic signaling pathways [4]. A common path to the EMT and gastrulation is the Wnt signaling pathway. Wnt plays an essential role in morphogenesis, cellular orientation, and organization. The pathway is activated at the posterior region of the embryo, resulting in the creation of the primitive streak. This is the same pathway that assists with the initiation of EMT. Later, after EMT facilitates the formation of the primitive streak, the opposite process, MET, gives rise to other components, such as the peripheral nervous system and adrenal medulla cells. Another interesting example is the formation of nephron epithelium in the kidney. Mesenchymal cells aggregate around the ureteral bud and acquire cell-to-cell adhesion properties by MET [6].
1.2. Type 2 EMT
The second type of EMT is associated with wound healing, tissue regeneration, and organ fibrosis. It begins as part of a regular healing event that typically creates fibroblasts and other cells to recreate tissues after trauma or an inflammatory process [3]. In contrast to type 1 EMT, these types are associated with inflammation and cease when it is attenuated. In chronic inflammation, the abnormal formation of myofibroblasts is associated with permanent progressive fibrosis, leading to organ failure due to high deposition of extracellular matrix components such as collagens, laminins, elastin, and tenascins [5]. Yin et al. showed that nintedanib (Ofev), a medication used nowadays mainly for the treatment of pulmonary fibrosis, may also be used for proliferative vitreoretinopathy (PVR) by preventing TGF-β2-induced EMT in retinal pigment epithelial cells and thus providing a new therapeutical target for PVR [7].
Few signaling pathways promote EMT in tumors, but nevertheless, it has been shown that these molecules, such as miRNA, ZEB, and TGF-β2, play a crucial role in abnormal scarring. Moreover, several studies showed the difference in miRNA expression between normal skin and hyperplastic scars, showing higher expression in the scarred tissue and a possible correlation with TGF-β2 signaling [5].
Until now, the number of medications and therapeutic possibilities for fibrosis is insufficient. For instance, there are only two FDA-approved drugs for pulmonary fibrosis (nintedanib and pirfenidone) [8]. In addition to natural substances that demonstrated the ability to alter the EMT, such as honey, curcumin, olea europea, and Paeonia lactiflora, tissue engineering is currently focusing on creating new treatment lines based on understanding the EMT process. One of the treatment possibilities suggested is polyacrylamide hydrogels, which are matrices with different properties and are known to modulate EMT. Such biomaterials possess different biochemical and biophysical properties, thus facilitating the physiological process of tissue regeneration [5].
1.3. Type 3 EMT
Type 3 EMT occurs in neoplastic cells that pass through genetic changes affecting oncogenes and tumor suppressor genes. Carcinoma cells undergoing this type of EMT may invade and metastasize and thus favor cancer progression. Features of EMT have been observed in various kinds of tumors, for example, in breast [4], prostate [9], ovarian [10], colon [11], and other cancers. Gavert et al. suggest a hypothesis, and according to it, developmental programs are reactivated during tumorigenesis, contributing to the overall cellular outcome. Many regulation checkpoints of the EMT process are highly expressed and are associated with a non-coordinated and less ordered process compared to the developmental EMT process [12].
Interestingly, an association was found between low expression of specific molecules such as E-cadherins and membranous β-catenin and the progression of prostate cancer. Furthermore, it has been shown that the EMT process, also known as cellular plasticity, is the cause of metastatic disease mediated by the cells that act as cancer stem cells [13]. Rusetti et al. showed on a mouse model that the cells that possess cellular plasticity are more prone to survive in distant regions other than the primary tumor, while those that did not pass through EMT could initiate a primary tumor but would not pass through further metastasis [14]. Jiaren Li et al. showed in different research that the accumulation of mesenchymal cells is strongly associated with benign prostatic hyperplasia (BPH). Therefore, he suggests using EMT as a potential target for treating BPH [15].
Other research showed, by protein expression analysis, that EMT in bone metastasis from prostate cancer creates cells that are often resistant to chemotherapeutic treatment due to their arrest in the G0 state while contacting bone marrow stromal cells. It has been shown that TGF-β, a potent activator of EMT and metastasis, plays a role in the induction of chemoresistance, as described [16]. In a work by Lu et al. on a 3D model system, it was found that TGF-β2 was highly induced in a co-culture with THP-1 cells. Hence, they tried to use an anti-TGF-β2 neutralizing antibody at various concentrations to determine the concentration that could inhibit macrophage-mediated growth of BPH-1. They found that anti-TGF-β2 neutralizing antibodies at a concentration of 50 ng/mL could inhibit the growth of BPH cells. Similarly, the anti-TGF-β2 neutralizing antibody attenuated the EMT induction marker (N-cadherin) expression. It increased the expression of E-cadherin, suggesting that TGF-β2 has a role in BPH formation, and neutralization of TGF-β2 may block the EMT that occurs in the pathogenesis of BPH [17].
2. Cellular and Tissue Morphology Characterizing EMT
Polarized epithelial cells, which are usually organized in stratified or single layers and acquire mesenchymal properties, have the capability of locomotion [18]. The fundamental events that impact the EMT process include a reduction in cell-cell adherence by repression and a change of location of cadherins, occludin, claudin, and desmoplakin [19] (Figure 1).
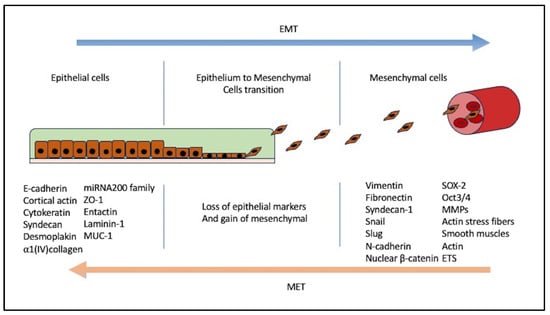
Figure 1. Epithelial mesenchymal transition. The process involves a transition of extracellular matrix components, anchoring the cell to the basement membrane. Losing epithelial anchors co-occurs with the gain of mesenchymal markers, indicating cells that have passed through EMT. Proteins that are downregulated during EMT are E-cadherin, β-catenin, laminin, desmoplakin, Muc-1, ZO-1, syndecan-1, cytokeratin 18, and the newly studied EPLIN [2]. Proteins gained are, for example, Snail, Twist, Slug, LEF-1, Scratch, SIP1, E47, Ets, FTS binding protein, RhoB, FSP1 (S100A4), TGF-beta, FGF-1,-2,-8, MMP-2, MMP-9, Vimentin, αSMA, FOXC2, fibronectin, collagen type 1, collagen type III, thrombospondin, PAI-1, etc. [2].
β-catenin is an E-cadherin-supporting molecule and frequently translocates from the cellular membrane to the nucleus to initiate further EMT events [20][21]. Circumferential F-actin fibers of the cytoskeleton are replaced by a network of stress fibers at the tips of which ECM adhesion molecules (including integrins, paxillin, and focal adhesion kinase) localize [22]. These changes allow cells to separate, leave the basement membrane, and lose the cellular apicobasal polarity, which is typical in epithelial cells, acquire a front-back polarity [23], and gain a different morphology, which is more variable, fibroblast-like cell shape for the facilitation of cell movement. Fibroblastoid cells play a role in the degradation or synthesis of ECM via different molecular pathways [18].
Tumor cells presenting the properties and the phenotype of mesenchymal cells show more invasiveness, metastatic capabilities, resistance to chemo and endocrine therapy, resistance to radiation-induced DNA damage, increased interaction with stromal inflammatory mechanisms, and increased cell survival [24][25].
A crucial part of the process is the change in the expression of various types of ECM components. For example, the expression of epithelial intermediate filaments such as β4 integrin and ZO-1 is reduced, while the equivalent mesenchymal proteins, such as vimentin, N-cadherin, and α-SMA, are typically increased. Matrix metalloproteases of different types (MMP-1, -2, -3, -7, and -14) are upregulated, thus allowing the cells to detach from each other and invade the basement membrane. The cells must undergo phenotypic changes to pass through the molecular modifications [26].
3. EMT Induction and Mechanism
EMT is induced by cytokines capable of proteolytic digestion of basement membranes on which the epithelial cell is attached. The process can be initiated by several oncogenes, including RASV12 [27], ErbB2 [28], and TRKB [29][30]. They activate many other components, such as PI3, MAP kinases, Notch, Wnt, and NF-κB pathways, all involved in EMT regulation [31]. Many other transcription factors, such as Snail 1, Snail 2, Twist, δEF1 (ZEB 1), SIP-1 (ZEB 2), and E12/E47, have been shown to induce the process of EMT. However, whether these factors function independently or combined with other elements to activate the EMT process is unknown.
Hierarchy exists in the expression of the factors. Snai1 is expressed in the early phases of the process, while other factors such as Snai2, Zeb1, and Twist are introduced later as part of the regulation and maintenance of migratory capabilities. Their role in EMT will be discussed later.
Growth factors such as TGF-β, EGF, IGF-II, FGF-2, and HGF are locally expressed and facilitate the process by binding epithelial receptors with specific intrinsic kinase activity. HGF, the ligand of the c-met receptor, was proclaimed in 1985 as the first EMT inducer molecule [32]. As a result, the tumor also starts to produce proteolytic enzymes. It remodels ECM and the basement membrane and prepares the environment to be suitable for migration and invasion.
Grant et al. describe that a hormonal-mediated axis regulates prostate cancer and that the early stages of the tumor’s proliferation are dependent on androgens. The tumor alters several signaling pathways for achieving invasive and aggressive capabilities, such as the Androgen receptor (AR) pathway and the EMT process. Therefore, many clinical trials and FDA-approved medications target the tumor via AR pathways, such as abiraterone acetate, a type of CYP17A1 inhibitor that blocks the synthesis of androgens, thus affecting tumor growth. The researchers emphasize that during embryogenesis, the prostate gland is formed and developmentally regulated by SRY-related high-mobility-group box (Sox) transcription factors. SOX9 is one of the factors which is shown to be significantly elevated in recurrent prostate cancer. Apparently, SOX9 takes part in Wnt/β-catenin and FGF signaling pathways that induce AR expression and EMT. The cells acquire the properties crucial for further metastasis [33]. Another interesting induction mechanism of EMT in prostate cancer is the heat shock protein (HSP).
An important induction mechanism was described by Lemster et al., according to which KDM5C modulates EMT signaling pathways such as Hedgehog, Wnt, Notch, PI3K-Akt-mTOR and other factors such as ZEB1, ZEB2, SNAI2, and others. KDM5C is a molecule that demethylates H3K4 histones. It has been shown that knockdown of KDM5C is associated with reduced risk for metastasis by high expression of E-cadherin. Also, KDM5C regulates the TGF-β signaling cascade by changes in SMAD7 expression. Therefore, inhibition of KDMC5 showed significant downregulation of SMAD7 and decreased TGF-β expression and EMT [34].
The TGF-β signaling pathway is also associated with converting fibroblasts into cancer-associated fibroblasts (CAF), which act as promoters of EMT. According to Wu et al., suppression of TGF-β by silibinin inhibits the differentiation of fibroblasts into CAF, suppresses the expression of vimentin and α-SMA, and thus, decreases the invasiveness of prostate cancer [35].
4. EMT Proteome and Genome; Molecular Switch
EMT proteome reflects the change in components, whether gained or lost, during the conversion of epithelial cells into mesenchymal cells in physiological cases or the transition of tumor epithelium to metastatic cells.
Studies on the EMT process are based mainly on the information acquired from proteins located in the epithelia rather than in fibroblasts or tumor cells. Many proteins are gained or maintained in the process and are described in Figure 1.
4.1. Twist
Twist is a transcription factor that plays an integral role in embryogenesis and is a crucial EMT regulator in cancers [36]. Twist and Snail1 upregulate Zeb1, leading to the downregulation of E-cadherin [37]. The ectopic expression of Twist in canine kidney cells caused a decrease in E-cadherin, α-, β- and γ-catenins, and therefore, acquirement of mesenchymal markers vimentin, fibronectin, smooth muscle actin, and N-cadherin [36].
Twits have been found to induce EMT in Hela and MCF7 cells, accompanied by the upregulation of ALDH1 and CD44, which are known as markers of stem cells [38]. In gastric cancer, the tumor stage and depth of invasion significantly correlate with the expression of Twist mRNA [39]. In vivo studies suggested that elevated Twist expression might be responsible for breast cancer lung metastases [36]. Moreover, it has been found that high expression of Twist is associated with high pre-treatment prostate-specific antigen (PSA) levels, high Gleason score (>7), advanced tumor stage, involvement of lymph nodes, distant metastasis, and biochemical progression. Also, a significant association was found between high Twist-1 and shorter biochemical progression-free survival [40].
In another article, it was described that the increased expression of Twist was in a primary tumor, while expression was reduced in distant metastasis. Also, a strong Twist expression was detected along with Slug and was associated with HIF-1α in localized prostate cancer, and a strong expression of twist was associated with HIF-1α in castration-resistant prostate cancer [41]. It was shown by Jin et al. that a protein known as trophinin-associated protein (TROAP) is overexpressed in prostate cancer and promotes its progression. At the same time, low levels can inhibit cancer cell proliferation and cause arrest in the cell cycle. Interestingly, they showed that TROAP knockdown induced apoptosis and prevented migration and invasion abilities by inhibiting the Twist/c-Myc pathway. On the other hand, overexpression of Twist showed a partial decrease in the inhibition of cellular proliferation induced by the silencing of TROAP [42].
4.2. Snail Family
Snails are essential for the induction of EMT. Snail1 is vital in forming the mesoderm layer and neural crest during embryogenesis and is an important factor in EMT-associated tumor progression. For example, Snail induces EMT in pancreatic cancer cells by expressing vimentin and suppressing E-cadherin. Moreover, Snail was found to be closely related to tumor growth properties, invasion, and metastasis [43].
In breast cancer, Snail1 is associated with tumor dedifferentiation. In invasive ductal carcinoma (IDC), Snail was observed in 47% of the cells and was expressed in most grade 3 tumors and more than half of grade 2 tumors but not in any grade 1 IDC [44]. The knockdown of Snail2 caused a full blockage of Twist1’s property to inhibit the transcription of E-cadherin [45]. Twist1 binds to a Snail 2 promoter to induce its transcription. The latter’s induction is essential for Twist 1-induced cell invasion and distant metastasis in mice. The expression of Twist 1 and Snail 2 is highly correlated in breast, bladder, and esophageal squamous cell carcinomas [44][46][47]. Gene expression analysis of CD44+/CD24− breast cells compared to CD44−/CD24+ cells revealed increased expression of 32 EMT-associated genes, including SLUG, ZEB-1, ZEB-2, periostin, Hedgehog signaling associated gene Gli-2, and the metastasis-associated gene SATB-1 [48]. Transgenic overexpression studies showed that only SLUG could change the phenotype of CD44−/CD24+ MCF-10A cells in favor of the induction of a subpopulation of CD44+/CD24− cells. In the luminal type of breast cancer cell line MCF-7, the overexpression of SLUG created cells with a CD44+/CD24+ phenotype, suggesting that basal cell types and not luminal types are more susceptible to acquisition of CD44+/CD24− phenotype, which is associated with EMT. Also, other specific EMT-associated genes inducing a CD44+/CD24− phenotype in MCF-10A cells are N-cadherin, FHL-1, ST-2, Wnt5B, FOXC2, ETV5, SATB-1, SLUG, and Gli-2. For example, it has been shown that the upregulation of the NF-κB subunit of p65 also upregulated ZEB-1 and ZEB-2 gene expression [49], which resulted in an increase in CD44+/CD24+ cells but not in CD44+/CD24- MCF-10A cells [48].
Katoh and Katoh have identified another member of the Snail family, Snail3. Human SNAI3 (Snail3) mRNA was expressed in skin melanoma, lung squamous cell carcinoma, and germ cell tumors. Authors have suggested that Snail/Gfi-1 (SNAG) zinc-finger proteins act as transcriptional repressors and are important in tumorigenesis and embryogenesis. Thus, it is suggested that these proteins, especially SNAI3, may be used as a promising and potent pharmacological target [50]. There is not enough experimental data on the Snail3 function [51].
Regarding prostate cancer, Snail has been associated with resistance to cisplatin and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), as there is a process of sensitization of the cells to cisplatin and TRAIL-mediated apoptosis. Moreover, the effect of Snail is an increase in migration and invasion. It has been shown on PTEN knockout mice with developed prostate cancer that there is a correlation between the expression of Snail and the stage of the tumor. Thus, there is low expression in benign prostatic metaplasia and high expression in bone metastatic specimens [51].
Snail overexpression correlates with the progression and disease stage of prostate cancer in vivo as well. Its activity is regulated by different growth factors such as TGF-β1, EGF, and vascular endothelial growth factor-A (VEGF-A). VEGF-A and TGF-β promote Snail nuclear localization in prostate cancer cells, while TGF-β and EGF induce EMT in prostate cancer cells, leading to their distinct metastatic potential. The mechanism by which VEGF-A contributes to the initiation of EMT is by binding to its receptor neuropilin-1 (NRP1), which then promotes nuclear localization of Snail. Surprisingly, there have been other factors that may express the overactivity of Snail, e.g., a hypoxic state. Hypoxia can influence EMT by regulation of VEGF-A expression and stimulation of the VEGF-A/NRP1 pathway that results in Snail nuclear localization and EMT in prostate cancer cells [51].
4.3. Zeb1 and Zeb2
The Zeb family consists of two essential members: Zeb1 (also known as TCF8 and δEF1) and Zeb2 (ZFXH1B and SIP1) [52]. The members of this family interact with the DNA by simultaneously binding the two zinc-finger domains to E-boxes [52]. Both proteins are potent repressors of CDH1 (E-cadherin gene). Although they are not as potent as Snail in the induction of EMT or in the repression of CDH1 in the in vitro assays [53], their silencing, especially that of Zeb1, has a higher impact on CDH1 expression than Snail [52][54]. Inhibition of Zeb1 and 2 by the miR200 family restores E-cadherin protein expression [55].
Snail1 and Slug activate Zeb family members, TCF3, TCF4, Twist, Goosecoid, and FOXC2, which may all be important in maintaining the EMT phenotype [56]. Docetaxel, a mitotic inhibitor chemotherapeutic, remains an essential line of treatment for advanced castration-resistant prostate cancer, but in some cases, resistance to the chemotherapeutic occurs. Hanrahan et al. showed an interesting connection between overexpression of EMT-related transcription factors from the ZEB family and the resistance of prostate cancer to Docetaxel. The repression of E-cadherins mediated the resistance and the EMT. When a knockdown of ZEB1 and 2 was performed, the E-cadherin expression increased markedly [57]; therefore, this pathway may be a potential line of treatment in docetaxel-resistant tumors.
4.4. MicroRNAs
Noncoding microRNAs are important signaling factors in EMT regulation [58]. MicroRNA 200 (miR200) and miR205 cause the inhibition of ZEB1 and ZEB2, which act as the repressors of E-cadherin and, thus, play a role in maintaining the epithelial cell phenotype [59]. In breast carcinoma, a loss of miR200 induces EMT via decreased E-cadherin and increased vimentin expression [59]. On the contrary, miR21 is upregulated in many other types of cancers and facilitates TGF-β–induced EMT [2]. It has been shown that miR200-mediated down-regulation of WAVE3, a metastatic favorable protein, significantly reduces the metastatic abilities of breast and prostate cancer cells [60]. A change in the cellular phenotype from mesenchymal to epithelial was achieved by inhibiting WAVE3 expression or by overexpression of miR200b or siRNA and indicates the role of WAVE3 in this process. Nevertheless, it is still unknown whether WAVE3 is directly involved in the ZEB1/ZEB2/E-cadherin pathway [60]. Sharma et al. explained that the molecular basis behind the overexpression of microRNA is the alteration in the copy number of microRNA, epigenetic modifications, upregulated expression of Dicer, a mutation in stem regions of pre-miRNA, single nucleotide polymorphism, and androgen receptor-regulated mechanisms [61].
Fabris et al. showed in a review that the microRNAs involved in almost any pathway and cellular process may be used as a potential marker in clinical practice. The fact that it can be found even in urine and semen samples that are highly correlated with the existence of prostate cancer opens a window to new diagnostic methods, especially since it has been suggested that both PSA and DRE (digital rectal examination) are neither specific nor sensitive enough. Fabris’s review showed a linear correlation between microRNA levels and cancer risk. The subtype of microRNA that was found to be constantly altered in the serum of prostate cancer patients is miR181 and is associated with EMT and apoptotic pathways. Interestingly, the review described miR96 as a marker that correlates with the survival of patients after radical prostatectomy and as a marker of biochemical recurrence, demonstrating that tumors expressing a high amount of miR96 have a significant decrease in recurrence-free survival [62]. Sekhon et al. showed that the loss of EMT-inhibiting microRNA or a gain of EMT-promoting microRNA leads to the induction of prostate cancer and to further progression, metastasis, and recurrence [63].
References
- Their, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454.
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784.
- Kalluri, R. EMT: When Epithelial Cells Decide to Become Mesenchymal-like Cells. J. Clin. Investig. 2009, 119, 1417–1419.
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression. J. Mammary Gland. Biol. Neoplasia 2010, 15, 117–134.
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587.
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2018, 7, 1.
- Yin, Y.; Liu, S.; Pu, L.; Luo, J.; Liu, H.; Wu, W. Nintedanib Prevents TGF-Β2-Induced Epithelial-Mesenchymal Transition in Retinal Pigment Epithelial Cells. Biomed. Pharmacother. 2023, 161, 114543.
- Salisbury, M.L.; Conoscenti, C.S.; Culver, D.A.; Yow, E.; Neely, M.L.; Bender, S.; Hartmann, N.; Palmer, S.M.; Leonard, T.B. Antifibrotic Drug Use in Patients with Idiopathic Pulmonary Fibrosis Data from the IPF-PRO Registry. Ann. Am. Thorac. Soc. 2020, 17, 1413–1423.
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F.H. Molecular Signature of Epithelial-Mesenchymal Transition (EMT) in Human Prostate Cancer Bone Metastasis. Am. J. Transl. Res. 2011, 3, 90.
- Vergara, D.; Merlot, B.; Lucot, J.P.; Collinet, P.; Vinatier, D.; Fournier, I.; Salzet, M. Epithelial-Mesenchymal Transition in Ovarian Cancer. Cancer Lett. 2010, 291, 59–66.
- Bates, R.C.; Pursell, B.M.; Mercurio, A.M. Epithelial-Mesenchymal Transition and Colorectal Cancer: Gaining Insights into Tumor Progression Using LIM 1863 Cells. Cells Tissues Organs 2007, 185, 29–39.
- Gavert, N.; Ben-Ze’ev, A. Epithelial-Mesenchymal Transition and the Invasive Potential of Tumors. Trends Mol. Med. 2008, 14, 199–209.
- Chaves, L.P.; Melo, C.M.; Saggioro, F.P.; Dos Reis, R.B.; Squire, J.A. Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics. Genes 2021, 12, 1900.
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759.
- Li, J.; Yao, H.; Huang, J.; Li, C.; Zhang, Y.; Xu, R.; Wang, Z.; Long, Z.; Tang, J.; Wang, L. METTL3 Promotes Prostatic Hyperplasia by Regulating PTEN Expression in an M6A-YTHDF2-Dependent Manner. Cell Death Dis. 2022, 13, 723.
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to Bone: A Fatal Attraction. Nat. Rev. Cancer 2011, 11, 411.
- Lu, T.; Lin, W.J.; Izumi, K.; Wang, X.; Xu, D.; Fang, L.Y.; Li, L.; Jiang, Q.; Jin, J.; Chang, C. Targeting Androgen Receptor to Suppress Macrophage-Induced EMT and Benign Prostatic Hyperplasia (BPH) Development. Mol. Endocrinol. 2012, 26, 1707–1715.
- Zavadil, J.; Böttinger, E.P. TGF-Beta and Epithelial-to-Mesenchymal Transitions. Oncogene 2005, 24, 5764–5774.
- Mathias, R.A.; Simpson, R.J. Towards Understanding Epithelial–Mesenchymal Transition: A Proteomics Perspective. Biochim. Biophys. Acta BBA Proteins Proteom. 2009, 1794, 1325–1331.
- Stemmer, V.; De Craene, B.; Berx, G.; Behrens, J. Snail Promotes Wnt Target Gene Expression and Interacts with Beta-Catenin. Oncogene 2008, 27, 5075–5080.
- Klymkowsky, M.W. β-Catenin and Its Regulatory Network. Hum. Pathol. 2005, 36, 225–227.
- Vignjevic, D.; Montagnac, G. Reorganisation of the Dendritic Actin Network during Cancer Cell Migration and Invasion. Semin. Cancer Biol. 2008, 18, 12–22.
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Acta Anat. 1995, 154, 8–20.
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of Homotypic Cell Adhesion by Epithelial-Mesenchymal Transition or Mutation Limits Sensitivity to Epidermal Growth Factor Receptor Inhibition. Mol. Cancer Ther. 2007, 6, 532–541.
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and Slug Mediate Radioresistance and Chemoresistance by Antagonizing P53-Mediated Apoptosis and Acquiring a Stem-like Phenotype in Ovarian Cancer Cells. Stem Cells 2009, 27, 2059–2068.
- Kharaishvili, G.; Bouchal, J. Extracellular Matrix Proteins and Epithelial Cell Plasticity in Progression of Breast and Prostate Cancer. Ph.D. Thesis, Palacky University, Olomouc, Czech Republic, 2011.
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGFβ Cooperatively Regulate Epithelial Cell Plasticity and Metastasis: Dissection of Ras Signaling Pathways. J. Cell Biol. 2002, 156, 299–313.
- Jenndahl, L.E.; Isakson, P.; Baeckström, D. C-ErbB2-Induced Epithelial-Mesenchymal Transition in Mammary Epithelial Cells Is Suppressed by Cell-Cell Contact and Initiated Prior to E-Cadherin Downregulation. Int. J. Oncol. 2005, 27, 439–448.
- Kupferman, M.E.; Jiffar, T.; El-Naggar, A.; Yilmaz, T.; Zhou, G.; Xie, T.; Feng, L.; Wang, J.; Holsinger, F.C.; Yu, D.; et al. TrkB Induces EMT and Has a Key Role in Invasion of Head and Neck Squamous Cell Carcinoma. Oncogene 2010, 29, 2047.
- Smit, M.A.; Geiger, T.R.; Song, J.-Y.; Gitelman, I.; Peeper, D.S. A Twist-Snail Axis Critical for TrkB-Induced Epithelial-Mesenchymal Transition-like Transformation, Anoikis Resistance, and Metastasis. Mol. Cell Biol. 2009, 29, 3722–3737.
- Smit, M.A.; Peeper, D.S. Epithelial-Mesenchymal Transition and Senescence: Two Cancer-Related Processes Are Crossing Paths. Aging 2010, 2, 735.
- Stoker, M.; Perryman, M. An Epithelial Scatter Factor Released by Embryo Fibroblasts. J. Cell Sci. 1985, 77, 209–223.
- Grant, C.M.; Kyprianou, N. Epithelial Mesenchymal Transition (EMT) in Prostate Growth and Tumor Progression. Transl. Androl. Urol. 2013, 2, 202–211.
- Lemster, A.L.; Sievers, E.; Pasternack, H.; Lazar-Karsten, P.; Klümper, N.; Sailer, V.; Offermann, A.; Brägelmann, J.; Perner, S.; Kirfel, J. Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT. Cancers 2022, 14, 1894.
- Wu, J.; Ji, H.; Li, T.; Guo, H.; Xu, H.; Zhu, J.; Tian, J.; Gao, M.; Wang, X.; Zhang, A. Targeting the Prostate Tumor Microenvironment by Plant-Derived Natural Products. Cell. Signal. 2024, 115, 111011.
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939.
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; De Herreros, A.G. Functional Cooperation between Snail1 and Twist in the Regulation of ZEB1 Expression during Epithelial to Mesenchymal Transition. J. Biol. Chem. 2011, 286, 12024–12032.
- Li, Q.; Chen, C.; Kapadia, A.; Zhou, Q.; Harper, M.K.; Schaack, J.; LaBarbera, D.V. 3D Models of Epithelial-Mesenchymal Transition in Breast Cancer Metastasis: High-Throughput Screening Assay Development, Validation, and Pilot Screen. J. Biomol. Screen. 2011, 16, 141–154.
- Otsuki, S.; Inokuchi, M.; Enjoji, M.; Ishikawa, T.; Takagi, Y.; Kato, K.; Yamada, H.; Kojima, K.; Sugihara, K. Vimentin Expression Is Associated with Decreased Survival in Gastric Cancer. Oncol. Rep. 2011, 25, 1235–1242.
- Abdelrahman, A.E.; Arafa, S.A.; Ahmed, R.A. Prognostic Value of Twist-1, E-Cadherin and EZH2 in Prostate Cancer: An Immunohistochemical Study. Turk. Patoloji Derg. 2017, 1, 198–210.
- Børretzen, A.; Gravdal, K.; Haukaas, S.A.; Mannelqvist, M.; Beisland, C.; Akslen, L.A.; Halvorsen, O.J. The Epithelial–Mesenchymal Transition Regulators Twist, Slug, and Snail Are Associated with Aggressive Tumour Features and Poor Outcome in Prostate Cancer Patients. J. Pathol. Clin. Res. 2021, 7, 253–270.
- Jin, L.; Zhou, Y.; Chen, G.; Dai, G.; Fu, K.; Yang, D.; Zhu, J. EZH2-TROAP Pathway Promotes Prostate Cancer Progression Via TWIST Signals. Front. Oncol. 2021, 10, 592239.
- Nishioka, R.; Itoh, S.; Gui, T.; Gai, Z.; Oikawa, K.; Kawai, M.; Tani, M.; Yamaue, H.; Muragaki, Y. SNAIL Induces Epithelial-to-Mesenchymal Transition in a Human Pancreatic Cancer Cell Line (BxPC3) and Promotes Distant Metastasis and Invasiveness in Vivo. Exp. Mol. Pathol. 2010, 89, 149–157.
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail Expression with Histological Grade and Lymph Node Status in Breast Carcinomas. Oncogene 2002, 21, 3241–3246.
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 Is an Essential Mediator of Twist1-Induced Epithelial-Mesenchymal Transition and Metastasis. Cancer Res. 2011, 71, 245.
- Yu, Q.; Zhang, K.; Wang, X.; Liu, X.; Zhang, Z. Expression of Transcription Factors Snail, Slug, and Twist in Human Bladder Carcinoma. J. Exp. Clin. Cancer Res. 2010, 29, 1–9.
- Xu, S.; Zhou, Y.; Biekemitoufu, H.; Wang, H.; Li, C.; Zhang, W.; Ma, Y. Expression of Twist, Slug and Snail in Esophageal Squamous Cell Carcinoma and Their Prognostic Significance. Oncol. Lett. 2021, 21, 184.
- Bhat-Nakshatri, P.; Appaiah, H.; Ballas, C.; Pick-Franke, P.; Goulet, R.; Badve, S.; Srour, E.F.; Nakshatri, H. SLUG/SNAI2 and Tumor Necrosis Factor Generate Breast Cells with CD44+/CD24− Phenotype. BMC Cancer 2010, 10, 411.
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-ΚB Represses E-Cadherin Expression and Enhances Epithelial to Mesenchymal Transition of Mammary Epithelial Cells: Potential Involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724.
- Katoh, M.; Katoh, M. Identification and Characterization of Human SNAIL3 (SNAI3) Gene in Silico. Int. J. Mol. Med. 2003, 11, 383–388.
- Smith, B.N.; Odero-Marah, V.A. The Role of Snail in Prostate Cancer. Cell Adhes. Migr. 2012, 6, 433–441.
- Vandewalle, C.; Van Roy, F.; Berx, G. The Role of the ZEB Family of Transcription Factors in Development and Disease. Cell. Mol. Life Sci. 2009, 66, 773–787.
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail Blocks the Cell Cycle and Confers Resistance to Cell Death. Genes. Dev. 2004, 18, 1131–1143.
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 Is a Transcriptional Repressor of E-Cadherin and Regulates Epithelial Plasticity in Breast Cancer Cells. Oncogene 2005, 24, 2375–2385.
- Kajita, M.; McClinic, K.N.; Wade, P.A. Aberrant Expression of the Transcription Factors Snail and Slug Alters the Response to Genotoxic Stress. Mol. Cell Biol. 2004, 24, 7559.
- Hugo, H.J.; Kokkinos, M.I.; Blick, T.; Ackland, M.L.; Thompson, E.W.; Newgreen, D.F. Defining the E-Cadherin Repressor Interactome in Epithelial-Mesenchymal Transition: The PMC42 Model as a Case Study. Cells Tissues Organs 2010, 193, 23–40.
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The Role of Epithelial-Mesenchymal Transition Drivers ZEB1 and ZEB2 in Mediating Docetaxel-Resistant Prostate Cancer. Mol. Oncol. 2017, 11, 251–265.
- Zavadil, J.; Narasimhan, M.; Blumenberg, M.; Schneider, R.J. Transforming Growth Factor-β and MicroRNA:MRNA Regulatory Networks in Epithelial Plasticity. Cells Tissues Organs 2007, 185, 157–161.
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601.
- Sossey-Alaoui, K.; Bialkowska, K.; Plow, E.F. The MiR200 Family of MicroRNAs Regulates WAVE3-Dependent Cancer Cell Invasion. J. Biol. Chem. 2009, 284, 33019–33029.
- Sharma, N.; Baruah, M.M. The MicroRNA Signatures: Aberrantly Expressed MiRNAs in Prostate Cancer. Clin. Transl. Oncol. 2019, 21, 126–144.
- Fabris, L.; Ceder, Y.; Chinnaiyan, A.M.; Jenster, G.W.; Sorensen, K.D.; Tomlins, S.; Visakorpi, T.; Calin, G.A. The Potential of MicroRNAs as Prostate Cancer Biomarkers. Eur. Urol. 2016, 70, 312–322.
- Sekhon, K.; Bucay, N.; Majid, S.; Dahiya, R.; Saini, S. MicroRNAs and Epithelial-Mesenchymal Transition in Prostate Cancer. Oncotarget 2016, 7, 67597–67611.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
763
Revisions:
2 times
(View History)
Update Date:
21 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No