Epithelial-mesenchymal transition (EMT) is a crucial and fundamental mechanism in many cellular processes, beginning with embryogenesis via tissue remodulation and wound healing, and plays a vital role in tumorigenesis and metastasis formation. EMT is a complex process that involves many transcription factors and genes that enable the tumor cell to leave the primary location, invade the basement membrane, and send metastasis to other tissues. Moreover, it may help the tumor avoid the immune system and establish radioresistance and chemoresistance. It may also change the normal microenvironment, thus promoting other key factors for tumor survival, such as hypoxia-induced factor-1 (HIF-1) and promoting neoangiogenesis.
- epithelial-mesenchymal transition
- EMT
- prostate cancer
- BPH
- transcription factors
1. Description of Types of EMT
1.1. Type 1 EMT
1.2. Type 2 EMT
1.3. Type 3 EMT
2. Cellular and Tissue Morphology Characterizing EMT
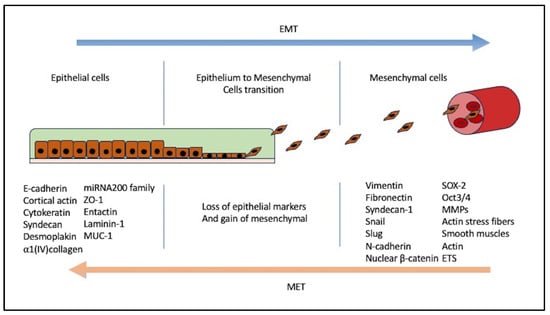
3. EMT Induction and Mechanism
4. EMT Proteome and Genome; Molecular Switch
4.1. Twist
4.2. Snail Family
4.3. Zeb1 and Zeb2
4.4. MicroRNAs
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines12020418
References
- Their, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454.
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784.
- Kalluri, R. EMT: When Epithelial Cells Decide to Become Mesenchymal-like Cells. J. Clin. Investig. 2009, 119, 1417–1419.
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression. J. Mammary Gland. Biol. Neoplasia 2010, 15, 117–134.
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587.
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2018, 7, 1.
- Yin, Y.; Liu, S.; Pu, L.; Luo, J.; Liu, H.; Wu, W. Nintedanib Prevents TGF-Β2-Induced Epithelial-Mesenchymal Transition in Retinal Pigment Epithelial Cells. Biomed. Pharmacother. 2023, 161, 114543.
- Salisbury, M.L.; Conoscenti, C.S.; Culver, D.A.; Yow, E.; Neely, M.L.; Bender, S.; Hartmann, N.; Palmer, S.M.; Leonard, T.B. Antifibrotic Drug Use in Patients with Idiopathic Pulmonary Fibrosis Data from the IPF-PRO Registry. Ann. Am. Thorac. Soc. 2020, 17, 1413–1423.
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F.H. Molecular Signature of Epithelial-Mesenchymal Transition (EMT) in Human Prostate Cancer Bone Metastasis. Am. J. Transl. Res. 2011, 3, 90.
- Vergara, D.; Merlot, B.; Lucot, J.P.; Collinet, P.; Vinatier, D.; Fournier, I.; Salzet, M. Epithelial-Mesenchymal Transition in Ovarian Cancer. Cancer Lett. 2010, 291, 59–66.
- Bates, R.C.; Pursell, B.M.; Mercurio, A.M. Epithelial-Mesenchymal Transition and Colorectal Cancer: Gaining Insights into Tumor Progression Using LIM 1863 Cells. Cells Tissues Organs 2007, 185, 29–39.
- Gavert, N.; Ben-Ze’ev, A. Epithelial-Mesenchymal Transition and the Invasive Potential of Tumors. Trends Mol. Med. 2008, 14, 199–209.
- Chaves, L.P.; Melo, C.M.; Saggioro, F.P.; Dos Reis, R.B.; Squire, J.A. Epithelial–Mesenchymal Transition Signaling and Prostate Cancer Stem Cells: Emerging Biomarkers and Opportunities for Precision Therapeutics. Genes 2021, 12, 1900.
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759.
- Li, J.; Yao, H.; Huang, J.; Li, C.; Zhang, Y.; Xu, R.; Wang, Z.; Long, Z.; Tang, J.; Wang, L. METTL3 Promotes Prostatic Hyperplasia by Regulating PTEN Expression in an M6A-YTHDF2-Dependent Manner. Cell Death Dis. 2022, 13, 723.
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to Bone: A Fatal Attraction. Nat. Rev. Cancer 2011, 11, 411.
- Lu, T.; Lin, W.J.; Izumi, K.; Wang, X.; Xu, D.; Fang, L.Y.; Li, L.; Jiang, Q.; Jin, J.; Chang, C. Targeting Androgen Receptor to Suppress Macrophage-Induced EMT and Benign Prostatic Hyperplasia (BPH) Development. Mol. Endocrinol. 2012, 26, 1707–1715.
- Zavadil, J.; Böttinger, E.P. TGF-Beta and Epithelial-to-Mesenchymal Transitions. Oncogene 2005, 24, 5764–5774.
- Mathias, R.A.; Simpson, R.J. Towards Understanding Epithelial–Mesenchymal Transition: A Proteomics Perspective. Biochim. Biophys. Acta BBA Proteins Proteom. 2009, 1794, 1325–1331.
- Stemmer, V.; De Craene, B.; Berx, G.; Behrens, J. Snail Promotes Wnt Target Gene Expression and Interacts with Beta-Catenin. Oncogene 2008, 27, 5075–5080.
- Klymkowsky, M.W. β-Catenin and Its Regulatory Network. Hum. Pathol. 2005, 36, 225–227.
- Vignjevic, D.; Montagnac, G. Reorganisation of the Dendritic Actin Network during Cancer Cell Migration and Invasion. Semin. Cancer Biol. 2008, 18, 12–22.
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Acta Anat. 1995, 154, 8–20.
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of Homotypic Cell Adhesion by Epithelial-Mesenchymal Transition or Mutation Limits Sensitivity to Epidermal Growth Factor Receptor Inhibition. Mol. Cancer Ther. 2007, 6, 532–541.
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and Slug Mediate Radioresistance and Chemoresistance by Antagonizing P53-Mediated Apoptosis and Acquiring a Stem-like Phenotype in Ovarian Cancer Cells. Stem Cells 2009, 27, 2059–2068.
- Kharaishvili, G.; Bouchal, J. Extracellular Matrix Proteins and Epithelial Cell Plasticity in Progression of Breast and Prostate Cancer. Ph.D. Thesis, Palacky University, Olomouc, Czech Republic, 2011.
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGFβ Cooperatively Regulate Epithelial Cell Plasticity and Metastasis: Dissection of Ras Signaling Pathways. J. Cell Biol. 2002, 156, 299–313.
- Jenndahl, L.E.; Isakson, P.; Baeckström, D. C-ErbB2-Induced Epithelial-Mesenchymal Transition in Mammary Epithelial Cells Is Suppressed by Cell-Cell Contact and Initiated Prior to E-Cadherin Downregulation. Int. J. Oncol. 2005, 27, 439–448.
- Kupferman, M.E.; Jiffar, T.; El-Naggar, A.; Yilmaz, T.; Zhou, G.; Xie, T.; Feng, L.; Wang, J.; Holsinger, F.C.; Yu, D.; et al. TrkB Induces EMT and Has a Key Role in Invasion of Head and Neck Squamous Cell Carcinoma. Oncogene 2010, 29, 2047.
- Smit, M.A.; Geiger, T.R.; Song, J.-Y.; Gitelman, I.; Peeper, D.S. A Twist-Snail Axis Critical for TrkB-Induced Epithelial-Mesenchymal Transition-like Transformation, Anoikis Resistance, and Metastasis. Mol. Cell Biol. 2009, 29, 3722–3737.
- Smit, M.A.; Peeper, D.S. Epithelial-Mesenchymal Transition and Senescence: Two Cancer-Related Processes Are Crossing Paths. Aging 2010, 2, 735.
- Stoker, M.; Perryman, M. An Epithelial Scatter Factor Released by Embryo Fibroblasts. J. Cell Sci. 1985, 77, 209–223.
- Grant, C.M.; Kyprianou, N. Epithelial Mesenchymal Transition (EMT) in Prostate Growth and Tumor Progression. Transl. Androl. Urol. 2013, 2, 202–211.
- Lemster, A.L.; Sievers, E.; Pasternack, H.; Lazar-Karsten, P.; Klümper, N.; Sailer, V.; Offermann, A.; Brägelmann, J.; Perner, S.; Kirfel, J. Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT. Cancers 2022, 14, 1894.
- Wu, J.; Ji, H.; Li, T.; Guo, H.; Xu, H.; Zhu, J.; Tian, J.; Gao, M.; Wang, X.; Zhang, A. Targeting the Prostate Tumor Microenvironment by Plant-Derived Natural Products. Cell. Signal. 2024, 115, 111011.
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939.
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; De Herreros, A.G. Functional Cooperation between Snail1 and Twist in the Regulation of ZEB1 Expression during Epithelial to Mesenchymal Transition. J. Biol. Chem. 2011, 286, 12024–12032.
- Li, Q.; Chen, C.; Kapadia, A.; Zhou, Q.; Harper, M.K.; Schaack, J.; LaBarbera, D.V. 3D Models of Epithelial-Mesenchymal Transition in Breast Cancer Metastasis: High-Throughput Screening Assay Development, Validation, and Pilot Screen. J. Biomol. Screen. 2011, 16, 141–154.
- Otsuki, S.; Inokuchi, M.; Enjoji, M.; Ishikawa, T.; Takagi, Y.; Kato, K.; Yamada, H.; Kojima, K.; Sugihara, K. Vimentin Expression Is Associated with Decreased Survival in Gastric Cancer. Oncol. Rep. 2011, 25, 1235–1242.
- Abdelrahman, A.E.; Arafa, S.A.; Ahmed, R.A. Prognostic Value of Twist-1, E-Cadherin and EZH2 in Prostate Cancer: An Immunohistochemical Study. Turk. Patoloji Derg. 2017, 1, 198–210.
- Børretzen, A.; Gravdal, K.; Haukaas, S.A.; Mannelqvist, M.; Beisland, C.; Akslen, L.A.; Halvorsen, O.J. The Epithelial–Mesenchymal Transition Regulators Twist, Slug, and Snail Are Associated with Aggressive Tumour Features and Poor Outcome in Prostate Cancer Patients. J. Pathol. Clin. Res. 2021, 7, 253–270.
- Jin, L.; Zhou, Y.; Chen, G.; Dai, G.; Fu, K.; Yang, D.; Zhu, J. EZH2-TROAP Pathway Promotes Prostate Cancer Progression Via TWIST Signals. Front. Oncol. 2021, 10, 592239.
- Nishioka, R.; Itoh, S.; Gui, T.; Gai, Z.; Oikawa, K.; Kawai, M.; Tani, M.; Yamaue, H.; Muragaki, Y. SNAIL Induces Epithelial-to-Mesenchymal Transition in a Human Pancreatic Cancer Cell Line (BxPC3) and Promotes Distant Metastasis and Invasiveness in Vivo. Exp. Mol. Pathol. 2010, 89, 149–157.
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail Expression with Histological Grade and Lymph Node Status in Breast Carcinomas. Oncogene 2002, 21, 3241–3246.
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 Is an Essential Mediator of Twist1-Induced Epithelial-Mesenchymal Transition and Metastasis. Cancer Res. 2011, 71, 245.
- Yu, Q.; Zhang, K.; Wang, X.; Liu, X.; Zhang, Z. Expression of Transcription Factors Snail, Slug, and Twist in Human Bladder Carcinoma. J. Exp. Clin. Cancer Res. 2010, 29, 1–9.
- Xu, S.; Zhou, Y.; Biekemitoufu, H.; Wang, H.; Li, C.; Zhang, W.; Ma, Y. Expression of Twist, Slug and Snail in Esophageal Squamous Cell Carcinoma and Their Prognostic Significance. Oncol. Lett. 2021, 21, 184.
- Bhat-Nakshatri, P.; Appaiah, H.; Ballas, C.; Pick-Franke, P.; Goulet, R.; Badve, S.; Srour, E.F.; Nakshatri, H. SLUG/SNAI2 and Tumor Necrosis Factor Generate Breast Cells with CD44+/CD24− Phenotype. BMC Cancer 2010, 10, 411.
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-ΚB Represses E-Cadherin Expression and Enhances Epithelial to Mesenchymal Transition of Mammary Epithelial Cells: Potential Involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724.
- Katoh, M.; Katoh, M. Identification and Characterization of Human SNAIL3 (SNAI3) Gene in Silico. Int. J. Mol. Med. 2003, 11, 383–388.
- Smith, B.N.; Odero-Marah, V.A. The Role of Snail in Prostate Cancer. Cell Adhes. Migr. 2012, 6, 433–441.
- Vandewalle, C.; Van Roy, F.; Berx, G. The Role of the ZEB Family of Transcription Factors in Development and Disease. Cell. Mol. Life Sci. 2009, 66, 773–787.
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail Blocks the Cell Cycle and Confers Resistance to Cell Death. Genes. Dev. 2004, 18, 1131–1143.
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 Is a Transcriptional Repressor of E-Cadherin and Regulates Epithelial Plasticity in Breast Cancer Cells. Oncogene 2005, 24, 2375–2385.
- Kajita, M.; McClinic, K.N.; Wade, P.A. Aberrant Expression of the Transcription Factors Snail and Slug Alters the Response to Genotoxic Stress. Mol. Cell Biol. 2004, 24, 7559.
- Hugo, H.J.; Kokkinos, M.I.; Blick, T.; Ackland, M.L.; Thompson, E.W.; Newgreen, D.F. Defining the E-Cadherin Repressor Interactome in Epithelial-Mesenchymal Transition: The PMC42 Model as a Case Study. Cells Tissues Organs 2010, 193, 23–40.
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The Role of Epithelial-Mesenchymal Transition Drivers ZEB1 and ZEB2 in Mediating Docetaxel-Resistant Prostate Cancer. Mol. Oncol. 2017, 11, 251–265.
- Zavadil, J.; Narasimhan, M.; Blumenberg, M.; Schneider, R.J. Transforming Growth Factor-β and MicroRNA:MRNA Regulatory Networks in Epithelial Plasticity. Cells Tissues Organs 2007, 185, 157–161.
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601.
- Sossey-Alaoui, K.; Bialkowska, K.; Plow, E.F. The MiR200 Family of MicroRNAs Regulates WAVE3-Dependent Cancer Cell Invasion. J. Biol. Chem. 2009, 284, 33019–33029.
- Sharma, N.; Baruah, M.M. The MicroRNA Signatures: Aberrantly Expressed MiRNAs in Prostate Cancer. Clin. Transl. Oncol. 2019, 21, 126–144.
- Fabris, L.; Ceder, Y.; Chinnaiyan, A.M.; Jenster, G.W.; Sorensen, K.D.; Tomlins, S.; Visakorpi, T.; Calin, G.A. The Potential of MicroRNAs as Prostate Cancer Biomarkers. Eur. Urol. 2016, 70, 312–322.
- Sekhon, K.; Bucay, N.; Majid, S.; Dahiya, R.; Saini, S. MicroRNAs and Epithelial-Mesenchymal Transition in Prostate Cancer. Oncotarget 2016, 7, 67597–67611.