 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Linsheng Zhang | -- | 2596 | 2024-02-19 13:34:11 | | | |
| 2 | Wendy Huang | Meta information modification | 2596 | 2024-02-20 03:42:54 | | |
Video Upload Options
Myelofibrosis refers to fibrosis in the bone marrow associated with certain bone marrow cancers. It is a characteristic of primary myelofibrosis and may develop later in other bone marrow cancers with overproduction of blood cells, such as polycythemia vera and essential thrombocythemia. It has been confirmed that mutations in three key genes, Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia oncogene (MPL), can increase the activity of blood-producing cells, make them grow more actively, and are associated with the development of myelofibrosis. Approximately 80% of myelofibrosis cases carry additional mutations that often involve proteins that control how genes are turned on and off. The presence of mutations provides evidence of a cancerous process. The order in which these mutations occur can influence how the disease manifests. Studies have shown that fibrosis is secondary to the cancerous process and is closely linked to abnormal cell growth driven by mutations.
1. Introduction
2. The Driver Mutations
3. Additional Mutations
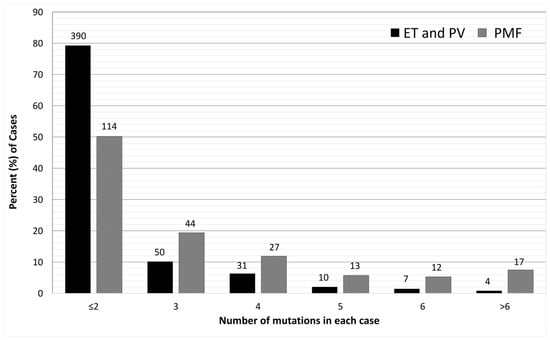
| Gene | Mutation Prevalence (%) | Most Frequent Mutations # | More Frequent in PMF Than Other MPN [34][43] | Clinical Relevance |
|---|---|---|---|---|
| Epigenetic Regulation (Chromosome Modification and DNA Methylation) | ||||
| ASXL1 | 21 | Truncation; E635Rfs | Yes | HMR Prevalence increases with age |
| DNMT3A | 12 | R882H/C | Yes | |
| EZH2 | 4 | Truncation and splice | Yes | HMR |
| IDH1/2 | 2 | IDH1 R132C/H, IDH2 R140Q/W | Yes | HMR Prevalence higher in other studies |
| TET2 | 17 | Truncation | No | The order of acquiring mutation affects phenotype |
| RNA splicing | ||||
| SF3B1 | 4 | K666N, K700E | No | Associated with ring sideroblasts |
| SRSF2 | 8 | P95 | Yes | HMR |
| U2AF1 | 5 | Q157, S34 | Yes | HMR |
| ZRSR2 | 2 | Truncation and splice | Yes | More common in SMF [41] |
| Signal transduction and transcription factors | ||||
| CBL | 6 | X366_splice, Y371H | No | Present with other additional mutations [44] Predict poor response to JAK inhibitors [45] |
| CUX1 | 3 | Truncation | Yes | |
| NFE2 | 2–5 * | E261fs | No, related to erythroid differentiation [25] | Associated with higher risk of transformation to AML, shorter OS. More common in SMF [41] |
| NRAS/KRAS | 9 | G12 | Yes | Relatively specific for MF [25][46] |
| RUNX1 | 4 | Truncation | Yes | Associated with transformation to AML [42] |
| SH2B3 | 1 | Truncation | No | May be considered a driver, or promoting JAK2 activity |
| TP53 | 2 | DNA-binding domain mutations | Yes | Relatively uncommon in MPNs. Associated with higher risk of transformation to AML [39]; however, low VAF in subclone may not increase risk [47] |
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and histiocytic/Dendritic neoplasms. Leukemia 2022, 36, 1703–1719.
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228.
- Passamonti, F.; Mora, B.; Barraco, D.; Maffioli, M. Post-ET and post-PV myelofibrosis: Updates on a distinct prognosis from primary myelofibrosis. Curr. Hematol. Malig. Rep. 2018, 13, 173–182.
- Puglianini, O.C.; Peker, D.; Zhang, L.; Papadantonakis, N. Essential thrombocythemia and post-essential thrombocythemia myelofibrosis: Updates on diagnosis, clinical aspects, and management. Lab. Med. 2023, 54, 13–22.
- Tefferi, A. How I treat myelofibrosis. Blood 2011, 117, 3494–3504.
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of myeloproliferative disorders. Annu. Rev. Pathol. 2016, 11, 101–126.
- Chen, M.-L.; Zhang, H.-C.; Yang, E.-P. Current status and hotspots evolution in myeloproliferative neoplasm: A bibliometric analysis from 2001 to 2022. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 4510–4519.
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Barosi, G.; Massa, M.; Campanelli, R.; Fois, G.; Catarsi, P.; Viarengo, G.; Villani, L.; Poletto, V.; Bosoni, T.; Magrini, U.; et al. Primary myelofibrosis: Older age and high JAK2V617F allele burden are associated with elevated plasma high-sensitivity C-reactive protein levels and a phenotype of progressive disease. Leuk. Res. 2017, 60, 18–23.
- Cazzola, M. Mutant calreticulin: When a chaperone becomes intrusive. Blood 2016, 127, 1219–1221.
- Kanagal-Shamanna, R.; Kikkeri, N.N.; Sandeep, S.D.; Mesa, R.; Giraudier, S.; Harrison, C.N.; Milojkovic, D. Primary Myelofibrosis. WHO Classifications of Tumours Online. Available online: https://tumourclassification.iarc.who.int/chaptercontent/63/15 (accessed on 3 December 2023).
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.-A.; Lee, D.S. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: Primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am. J. Clin. Pathol. 2015, 143, 635–644.
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468.
- Pecquet, C.; Papadopoulos, N.; Balligand, T.; Chachoua, I.; Tisserand, A.; Vertenoeil, G.; Nédélec, A.; Vertommen, D.; Roy, A.; Marty, C.; et al. Secreted mutant calreticulins as rogue cytokines in myeloproliferative neoplasms. Blood 2023, 141, 917–929.
- Guglielmelli, P.; Maccari, C.; Sordi, B.; Balliu, M.; Atanasio, A.; Mannarelli, C.; Capecchi, G.; Sestini, I.; Coltro, G.; Loscocco, G.G.; et al. Phenotypic correlations of CALR mutation variant allele frequency in patients with myelofibrosis. Blood Cancer J. 2023, 13, 21.
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477.
- Wang, Y.; Fan, D.; Zhang, X. Overt primary myelofibrosis with coexistence of JAK2V617F and MPLW515R driver mutations. Int. J. Lab. Hematol. 2024, 46, 180–182.
- Wang, Y.; Ran, F.; Lin, J.; Zhang, J.; Ma, D. Genetic and clinical characteristics of patients with Philadelphia-negative myeloproliferative neoplasm carrying concurrent mutations in JAK2V617F, CALR, and MPL. Technol. Cancer Res. Treat. 2023, 22, 15330338231154092.
- Thompson, E.R.; Nguyen, T.; Kankanige, Y.; Yeh, P.; Ingbritsen, M.; McBean, M.; Semple, T.; Mir Arnau, G.; Burbury, K.; Lee, N.; et al. Clonal independence of JAK2 and CALR or MPL mutations in comutated myeloproliferative neoplasms demonstrated by single cell DNA sequencing. Haematologica 2021, 106, 313–315.
- Pennisi, M.S.; Di Gregorio, S.; Tirrò, E.; Romano, C.; Duminuco, A.; Garibaldi, B.; Giuffrida, G.; Manzella, L.; Vigneri, P.; Palumbo, G.A. Additional genetic alterations and clonal evolution of MPNs with double mutations on the MPL gene: Two case reports. Hematol. Rep. 2023, 15, 317–324.
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332.
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342.
- Chang, Y.-C.; Lin, H.-C.; Chiang, Y.-H.; Chen, C.G.-S.; Huang, L.; Wang, W.-T.; Cheng, C.-C.; Lin, J.; Chang, Y.-F.; Chang, M.-C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 83.
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430.
- Lemoine, S.; Mornet, C.; Quintin-Roue, I.; Rousselet, M.-C.; Cottin, L.; Georgeais, A.; Dubouis, L.; Boyer, F.; Orvain, C.; Caillon, C.; et al. Histological and genetic characterization and follow-up of 130 patients with chronic triple-negative thrombocytosis. Haematologica 2022, 107, 2725–2731.
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.-J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111.
- Alduaij, W.; McNamara, C.J.; Schuh, A.; Arruda, A.; Sukhai, M.; Kanwar, N.; Thomas, M.; Spiegel, J.; Kennedy, J.A.; Stockley, T.; et al. Clinical utility of next-generation sequencing in the management of myeloproliferative neoplasms: A single-center experience. Hemasphere 2018, 2, e44.
- Delic, S.; Rose, D.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426.
- AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831.
- Pastor-Galán, I.; Martín, I.; Ferrer, B.; Hernández-Boluda, J.-C. Impact of molecular profiling on the management of patients with myelofibrosis. Cancer Treat. Rev. 2022, 109, 102435.
- Mroczkowska-Bękarciak, A.; Wróbel, T. BCR::ABL1-negative myeloproliferative neoplasms in the era of next-generation sequencing. Front. Genet. 2023, 14, 1241912.
- Hussein, K.; Van Dyke, D.L.; Tefferi, A. Conventional cytogenetics in myelofibrosis: Literature review and discussion. Eur. J. Haematol. 2009, 82, 329–338.
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the mutational profiles of primary myelofibrosis, polycythemia vera, and essential thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452.
- Rolles, B.; Mullally, A. Molecular pathogenesis of myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 319–329.
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228.
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451.
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer 2020, 59, 30–39.
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451.
- Yan, X.; Xu, Z.; Zhang, P.; Sun, Q.; Jia, Y.; Qin, T.; Qu, S.; Pan, L.; Li, Z.; Liu, J.; et al. Non-driver mutations landscape in different stages of primary myelofibrosis determined ASXL1 mutations play a critical role in disease progression. Blood Cancer J. 2023, 13, 56.
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921.
- Pettersson, H.; Adamsson, J.; Johansson, P.; Nilsson, S.; Palmqvist, L.; Andréasson, B.; Asp, J. The clinical relevance of broad mutational screening of myeloproliferative neoplasms at diagnosis. Front. Oncol. 2023, 13, 1190305.
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687.
- Reynolds, S.B.; Pettit, K.; Kandarpa, M.; Talpaz, M.; Li, Q. Exploring the molecular landscape of myelofibrosis, with a focus on Ras and mitogen-activated protein (MAP) kinase signaling. Cancers 2023, 15, 4654.
- Rodriguez-Meira, A.; Rahman, H.; Norfo, R.; Wen, W.; Chédeville, A.; O’Sullivan, J.; Wang, G.; Paterson, A.; Louka, E.; Brierley, C.K.; et al. Single-cell multi-omics reveals the genetic, cellular and molecular landscape of TP53 mutated leukemic transformation in MPN. Blood 2021, 138, 3.
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867.
- Marcault, C.; Zhao, L.-P.; Maslah, N.; Verger, E.; Daltro de Oliveira, R.; Soret-Dulphy, J.; Cazaux, M.; Gauthier, N.; Roux, B.; Clappier, E.; et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms. Blood 2021, 138, 2142–2148.
- Chifotides, H.T.; Verstovsek, S.; Bose, P. Association of myelofibrosis phenotypes with clinical manifestations, molecular profiles, and treatments. Cancers 2023, 15, 3331.
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496.
- Nielsen, H.M.; Andersen, C.L.; Westman, M.; Kristensen, L.S.; Asmar, F.; Kruse, T.A.; Thomassen, M.; Larsen, T.S.; Skov, V.; Hansen, L.L.; et al. Epigenetic changes in myelofibrosis: Distinct methylation changes in the myeloid compartments and in cases with ASXL1 mutations. Sci. Rep. 2017, 7, 6774.
- Nam, A.S.; Kim, K.-T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360.
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol. Cell 2019, 73, 1292–1305.
- Willekens, C.; Laplane, L.; Dagher, T.; Benlabiod, C.; Papadopoulos, N.; Lacout, C.; Rameau, P.; Catelain, C.; Alfaro, A.; Edmond, V.; et al. SRSF2-P95H decreases JAK/STAT signaling in hematopoietic cells and delays myelofibrosis development in mice. Leukemia 2023, 37, 1287–1297.
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.-S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379.
- Maslah, N.; Verger, E.; Giraudier, S.; Chea, M.; Hoffman, R.; Mascarenhas, J.; Cassinat, B.; Kiladjian, J.-J. Single-cell analysis reveals selection of TP53-mutated clones after MDM2 inhibition. Blood Adv. 2022, 6, 2813–2823.
- Gagelmann, N.; Badbaran, A.; Salit, R.B.; Schroeder, T.; Gurnari, C.; Pagliuca, S.; Panagiota, V.; Rautenberg, C.; Cassinat, B.; Thol, F.; et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood 2023, 141, 2901–2911.
- Dilip, D.; Menghrajani, K.; Glass, J.; Rampal, R.K.; Famulare, C.; Sirenko, M.; Levine, R.L.; Koche, R. MPN transformation is characterized by heterogeneous shifts in lineage character. Blood 2023, 142 (Suppl. 1), 749.

