Myelofibrosis refers to fibrosis in the bone marrow associated with certain bone marrow cancers. It is a characteristic of primary myelofibrosis and may develop later in other bone marrow cancers with overproduction of blood cells, such as polycythemia vera and essential thrombocythemia. It has been confirmed that mutations in three key genes, Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia oncogene (MPL), can increase the activity of blood-producing cells, make them grow more actively, and are associated with the development of myelofibrosis. Approximately 80% of myelofibrosis cases carry additional mutations that often involve proteins that control how genes are turned on and off. The presence of mutations provides evidence of a cancerous process. The order in which these mutations occur can influence how the disease manifests. Studies have shown that fibrosis is secondary to the cancerous process and is closely linked to abnormal cell growth driven by mutations.
- myelofibrosis
- driver mutations
- additional mutations
- myeloid neoplasms
- JAK2
- CALR
- MPL
1. Introduction
2. The Driver Mutations
The discovery of recurrent mutations in Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia oncogene (MPL) as driver mutations has transformed the diagnostic approach of MPN, as evident in the revisions of WHO classifications [1][8][9][1,8,9]. Clonal evidence, supported by the presence of a driver or other mutations commonly associated with various myeloid neoplasms, is crucial for definitive diagnosis. Both JAK2 and MPL encode proteins that activate the JAK/STAT signaling pathway, which is essential for signal transduction from erythropoietin (EPO), thrombopoietin (TPO), and granulocyte colony-stimulating factor (G-CSF) receptors. The pathobiology and diagnostic relevance of activating mutations in JAK2, CALR, and MPL have been extensively investigated in the clinical setting. JAK2 V617F mutation is associated with an increased risk of thrombosis, and a high allele burden is associated with disease progression [10]. MPL encodes the TPO receptor, and mutations, usually at codon W515, lead to constitutively active signaling independent of ligand binding. The interaction between MPL and altered calreticulin encoded by mutant CALR results in MPL hyperactivity [11]. CALR and MPL mutations are typically exclusive to ET and PMF and very rarely occur in PV [10]; however, JAK2 V617F mutation remains the most common driver mutation in PMF, reported in 50–60% of cases, followed by CALR mutations in 25–35% and MPL mutations in 5–10% cases [12][13][12,13]. Interestingly, JAK2 exon 12 mutations [14], which are also activating mutations, have not been documented in ET or PMF. All oncogenic CALR mutations are frame-shifting insertions or deletions (indels) that alter the C-terminal end of calreticulin from negatively charged acidic amino acids, aspartic acid (D)- and glutamic acid (E)-rich, to positively charged basic amino acids, arginine (R)- and lysine (K)-rich, removing the endoplasmic reticulin retention signal KDEL. Mutant calreticulin can be secreted and functions as a cytokine, retaining its ability to bind to MPL in the CALR-mutated clone [15]. In PMF, type 1 CALR mutations (51 bp deletion, L367Tfs*46) are approximately three times more prevalent than type 2 (5 bp insertion, K385Nfs*47) [13], with phenotypic variations observed among CALR mutation types [16]. In PMF, type 1 CALR mutations correlate with lower leukocytosis, lower bone marrow cellularity, and an increased number of megakaryocytes [13], while type 2 mutations align more closely with the phenotype of cases harboring JAK2 V617F [17]. In the vast majority of MPN cases, driver mutations in JAK2, CALR, and MPL are mutually exclusive. However, there have been occasional reports of cases exhibiting coexistence of JAK2 V617F, MPL, and/or CALR mutations [18][19][18,19]. Such cases likely involve distinct subclones of neoplastic cells harboring different driver mutations, as demonstrated by a single-cell sequencing study [20], although instances of dual mutations in a single clone have also been documented [21]. Approximately 10% of MPN cases lack detectable canonical mutations in JAK2, CALR, or MPL, categorizing them as triple-negative (TN) MPNs. A small subset of these cases may not truly be TN, as other rare gain-of-function mutations in one of these three genes, particularly MPL, have been reported [22][23][24][25][26][22,23,24,25,26]. True TN cases often harbor mutations outside of these three genes, confirming clonal hematopoiesis. However, these mutations, which are also prevalent in other myeloid neoplasms, are not considered driver mutations of MPNs. Despite the availability of NGS tests for clinical analysis, the driver mutations of TN cases have not yet been determined, even with comprehensive whole-exome sequencing (WES) studies. One candidate driver, SH2B3 mutation, has been identified in a subset of TN MPNs [27]. However, SH2B3 mutations and other driver mutations are not mutually exclusive. The pathogenic drivers of TN MPNs are either heterogeneous non-recurrent mutations, more complicated alterations that evade ready identification by currently available methods, or with mechanisms not yet recognized. Further exploration to understand the regulatory sequences within the non-coding regions of the human genome may shed light on the drivers and molecular pathogenesis of TN MPNs.3. Additional Mutations
With the accumulation of mutation profiling data from clinical studies, it is now clear that over 50% patients with MPNs harbor mutations in addition to driver mutations. Among the classic MPNs, PMF has the highest prevalence of additional mutations. With targeted sequencing of myeloid neoplasm-related genes, additional mutations have been reported in approximately 50% of PV and ET cases, and as high as 80% of PMF cases [28][29][30][28,29,30]. PMF also harbors a higher number of mutations than PV or ET [28][29][31][28,29,31] (Figure 2). Although additional mutations are not considered driver mutations of MPNs, they help establish the clonal nature of TN patients and have been integrated into the major diagnostic criteria of MPNs [1][2][1,2]. A query of the American Association for Cancer Research (AACR) Project GENIE public database in cBioportal [32] found 299 samples from 202 cases documented as PMF (https://genie.cbioportal.org/study?id=6562046bb01fff74fbb6c576 (accessed on 25 November 2023)). In these 299 samples, in addition to JAK2 (44.8%), CALR (14.7%), and MPL (9.4%) mutations, the prevalence of other mutations is similar to those reported by other studies [29][31][33][34][29,31,33,34]. Table 1 lists the prevalence of relatively frequent non-driver mutations and the most common mutations or mutation types cataloged in the GENIE database. In addition to the mutations detected in sequencing studies, cytogenetic abnormalities have been reported in 30–57% of PMF cases. However, none of the abnormal karyotypes are specific to PMF [35].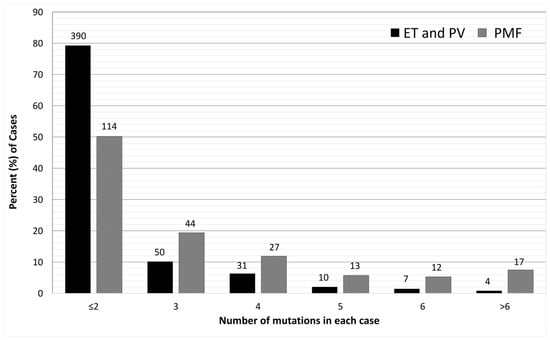
| Gene | Mutation Prevalence (%) | Most Frequent Mutations # | More Frequent in PMF Than Other MPN [34][43][34,43] | Clinical Relevance |
|---|---|---|---|---|
| Epigenetic Regulation (Chromosome Modification and DNA Methylation) | ||||
| ASXL1 | 21 | Truncation; E635Rfs | Yes | HMR Prevalence increases with age |
| DNMT3A | 12 | R882H/C | Yes | |
| EZH2 | 4 | Truncation and splice | Yes | HMR |
| IDH1/2 | 2 | IDH1 R132C/H, IDH2 R140Q/W | Yes | HMR Prevalence higher in other studies |
| TET2 | 17 | Truncation | No | The order of acquiring mutation affects phenotype |
| RNA splicing | ||||
| SF3B1 | 4 | K666N, K700E | No | Associated with ring sideroblasts |
| SRSF2 | 8 | P95 | Yes | HMR |
| U2AF1 | 5 | Q157, S34 | Yes | HMR |
| ZRSR2 | 2 | Truncation and splice | Yes | More common in SMF [41] |
| Signal transduction and transcription factors | ||||
| CBL | 6 | X366_splice, Y371H | No | Present with other additional mutations [44] Predict poor response to JAK inhibitors [45] |
| CUX1 | 3 | Truncation | Yes | |
| NFE2 | 2–5 * | E261fs | No, related to erythroid differentiation [25] | Associated with higher risk of transformation to AML, shorter OS. More common in SMF [41] |
| NRAS/KRAS | 9 | G12 | Yes | Relatively specific for MF [25][46][25,46] |
| RUNX1 | 4 | Truncation | Yes | Associated with transformation to AML [42] |
| SH2B3 | 1 | Truncation | No | May be considered a driver, or promoting JAK2 activity |
| TP53 | 2 | DNA-binding domain mutations | Yes | Relatively uncommon in MPNs. Associated with higher risk of transformation to AML [39]; however, low VAF in subclone may not increase risk [47] |