Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Miquéias Lopes-Pacheco | -- | 2676 | 2024-02-13 14:01:48 | | | |
| 2 | Catherine Yang | Meta information modification | 2676 | 2024-02-17 04:19:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ferreira, F.C.; Buarque, C.D.; Lopes-Pacheco, M. Ivacaftor (VX-770). Encyclopedia. Available online: https://encyclopedia.pub/entry/55020 (accessed on 21 July 2026).
Ferreira FC, Buarque CD, Lopes-Pacheco M. Ivacaftor (VX-770). Encyclopedia. Available at: https://encyclopedia.pub/entry/55020. Accessed July 21, 2026.
Ferreira, Filipa C., Camilla D. Buarque, Miquéias Lopes-Pacheco. "Ivacaftor (VX-770)" Encyclopedia, https://encyclopedia.pub/entry/55020 (accessed July 21, 2026).
Ferreira, F.C., Buarque, C.D., & Lopes-Pacheco, M. (2024, February 13). Ivacaftor (VX-770). In Encyclopedia. https://encyclopedia.pub/entry/55020
Ferreira, Filipa C., et al. "Ivacaftor (VX-770)." Encyclopedia. Web. 13 February, 2024.
Copy Citation
Ivacaftor possesses a molecular structure characterized by the presence of an N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydro-quinoline-3-carboxamide moiety. The quinolone scaffold within its composition is a crucial pharmacophore, significantly influencing drug discovery. This scaffold holds prominence as one of the primary classes of nitrogen-containing heterocycles found in various biologically active compounds and blockbuster drugs, as highlighted in the literature. The amide group serves as a crucial link between the “privileged building block” and the di-tert-butylphenol in ivacaftor’s structure. This linkage is of considerable importance in medicinal chemistry due to its multifaceted role.
CFTR potentiator
cystic fibrosis
1. Synthetic Routes
In recent years, several synthetic routes have been explored for ivacaftor, primarily focusing on the synthesis of dihydroquinoline fragments 4a or 4b (Scheme 1). These fragments are subsequently connected to 5-amino-2,4-di-tert-butyl-phenol (5a) or its protected derivative 5b through amide bond coupling, as outlined in approaches A to D. An alternative method involves the synthesis of the quinoline moiety in the final stages, commencing with methyl anthranilate, (E)-3-methoxyacryloyl chloride, and 5-amino-2,4-di-tert-butylphenyl methyl carbonate (5b) (approach E) [1].
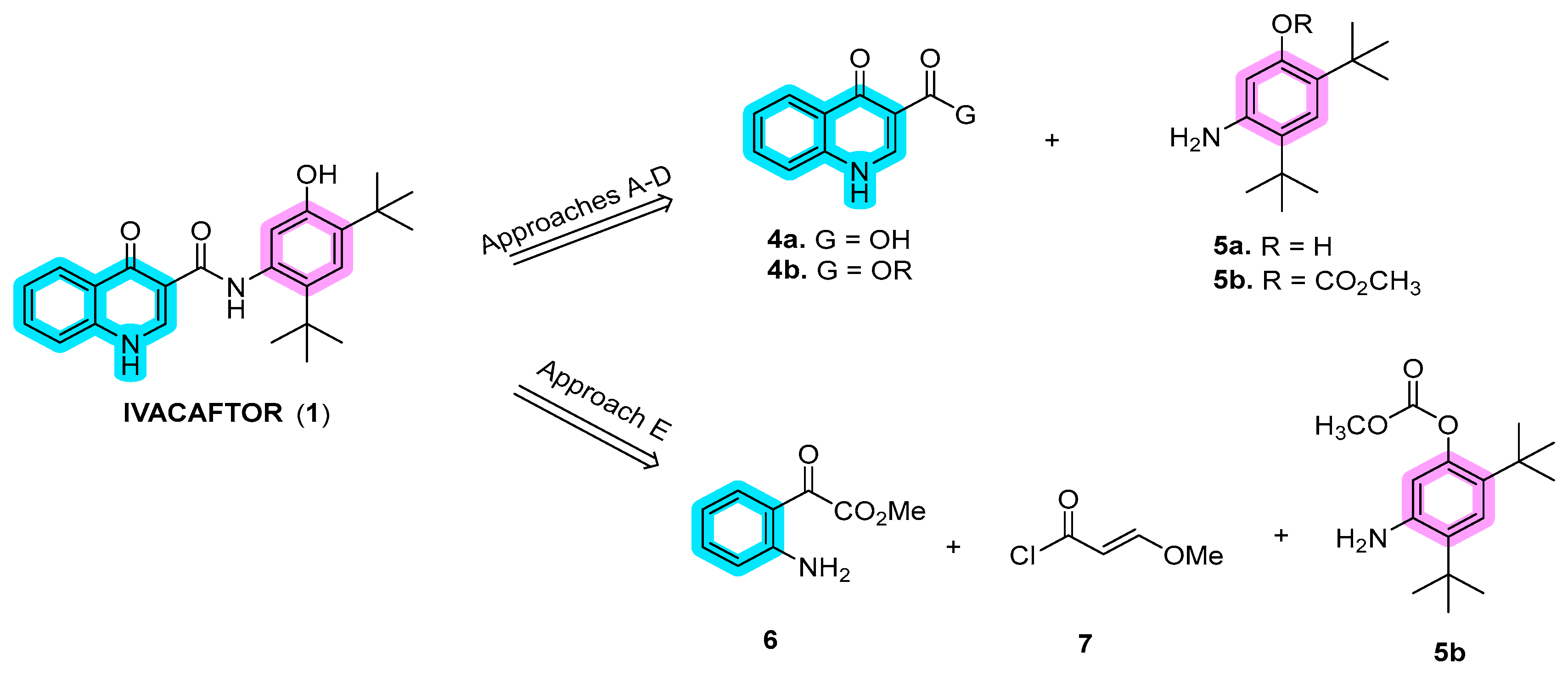
Scheme 1. Disconnection approaches for the synthesis of ivacaftor (1).
In the first stage of synthesizing quinoline moiety 4 (approaches A–D), Vertex introduced the Gould–Jacobs reaction using diethyl ethoxymethylene malonate (8) and aniline (9), as documented in a pivotal work [2]. Vertex has adapted and modified this route over the years to meet the demands of large-scale production [3][4]. Approaches B and C share similarities and were independently developed by the Shanghai University of Engineering Science [5] and Laurus Pharma [6]. These results are outlined in Scheme 2. Yang’s approach started from o-nitrobenzoyl acid (10a), while Laurus Pharma initiated the synthesis with o-fluoro-benzoyl acid (10b). In approach D, Vasudevan and co-authors reported the synthesis of ivacaftor using the Witkop–Winterfeldt oxidation of indolyl group 14 to form the quinoline through ozone oxidation [7][8]. For amide bond coupling with quinoline, Vertex employed aniline 5a in the initial route or 5-amino-2,4-di-tert-butylphenyl methyl carbonate (5b) in the second improved route, both derived from di-tert-butyl-phenol (15). In approach B, only aniline 5a was utilized [9], whereas in approach E, 5b was used instead. This intricate synthesis strategy showcases the diverse approaches taken by various researchers in the development of ivacaftor.
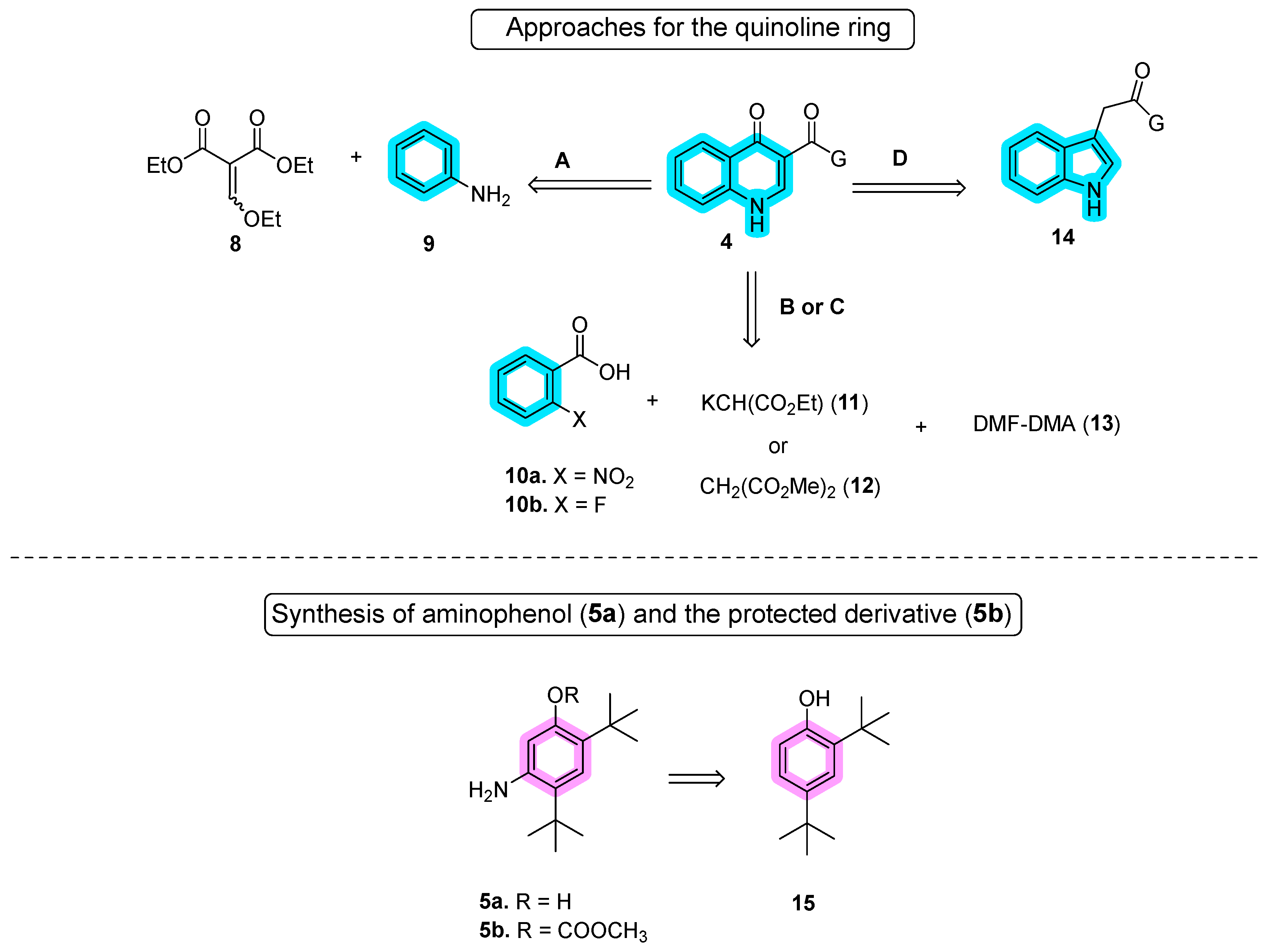
Scheme 2. Different approaches for the synthesis of quinoline and aniline moieties.
Approach A, as employed by Vertex in the synthesis of ivacaftor (Scheme 3), is noteworthy for its efficiency. In the initial report of this approach, quinoline 4a was synthesized through a three-step convergent route utilizing the Gould–Jacobs reaction, while aniline 5a was obtained in four steps, as detailed below. The pivotal connection of these building blocks occurred through amide bonding coupling using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), in dimethylformamide (DMF). The reaction typically involves the activation of a carboxylic acid group to form an active intermediate, which then reacts with an amine to produce the amide bond with a high coupling efficiency in relatively mild reaction conditions. Subsequent purification by column chromatography resulted in ivacaftor with a notable 71% yield [10][11][12][13].
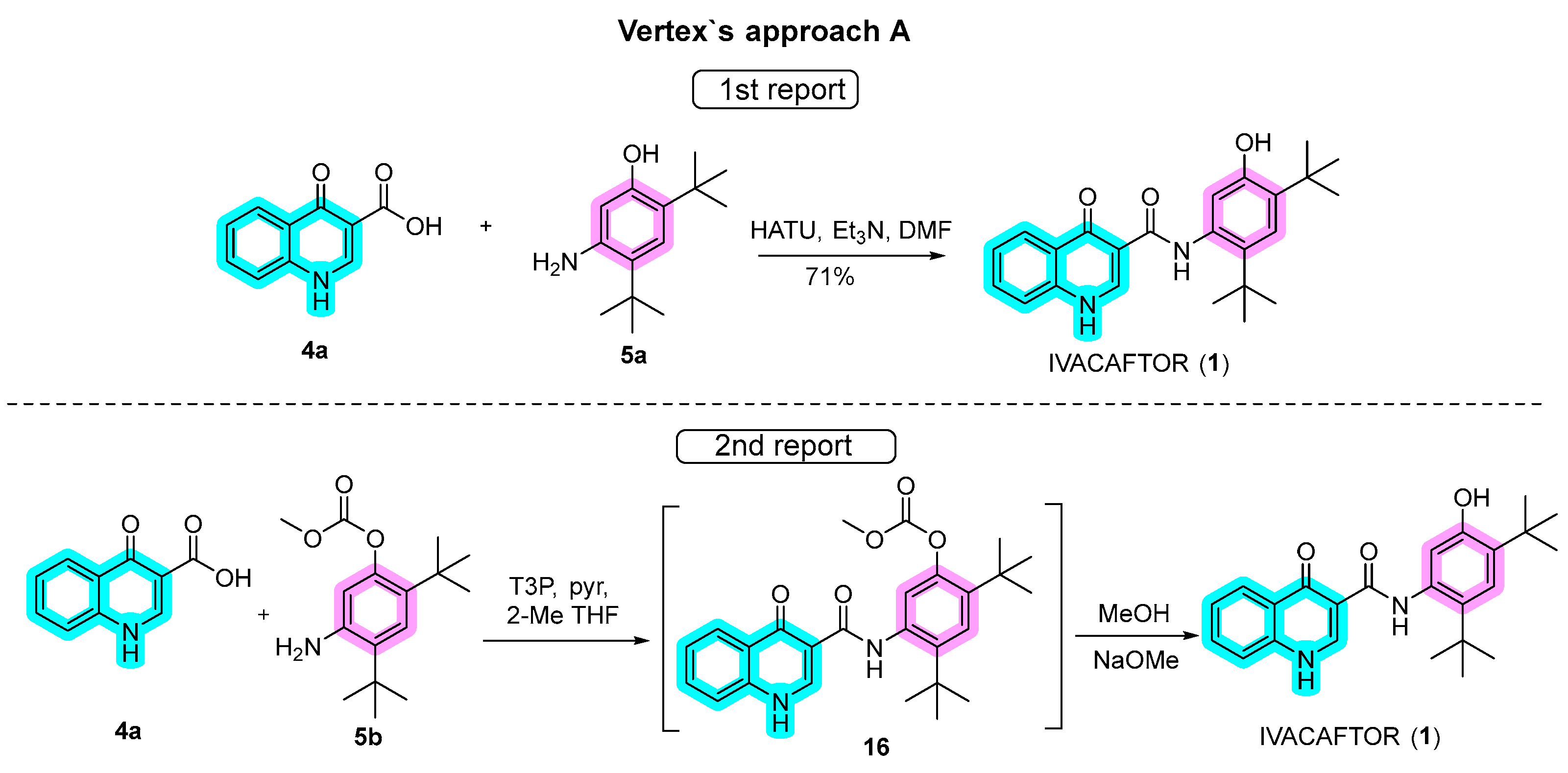
Scheme 3. Approaches for the synthesis of ivacaftor used by Vertex.
In a subsequent report, refinements to the synthetic route were implemented. Protected aniline 5b replaced 5a, and amide coupling was facilitated by propanephosphonic acid anhydride (T3P) with pyridine in 2-methyltetrahydrofuran (2-MeTHF), an alternative to HATU. While the reason for the switch to T3P as a coupling agent is not evident, it exhibits effectiveness under relatively mild conditions, reducing the risk of side reactions or unwanted byproducts. Additionally, it is compatible with a wide range of functional groups, rendering it suitable for use with a variety of substrates. Furthermore, T3P is a crystalline solid, which generally makes it easy to handle and store. Despite an additional protection step, intermediate 16 was conveniently hydrolyzed to ivacaftor in the same pot by the addition of MeOH and MeONa. A notable advantage of this modified approach was the elimination of chromatography steps, although specific yields for each step were not provided.
Beyond the choice of amide coupling and/or the use of protected or deprotected aniline in the final step, notable advancements have been made in the synthesis of quinoline 4b and aniline 5a or its derivative 5b in recent years (Scheme 4). For quinoline 4b, the Gould–Jacobs reaction played a crucial role in generating quinolone 4a. This involved a Claisen condensation between aniline 9 and diethyl ethoxymethylene malonate 8, followed by Friedel−Crafts cycloacylation. The methodologies differed in some aspects. In the initial approach, diethyl ethoxymethylene malonate 8 reacted with aniline 9 under neat conditions for 2 h at 140–150 °C to yield enamine 18. Subsequently, a POCl3/polyphosphoric acid (PPA) mixture was employed for the Friedel–Crafts cycloacylation through intermediates 19 and 20 [14], resulting in a 64% yield over the three steps. The mixture of POCl3/PPA is important to facilitate the cyclization process. POCl3 may potentially convert carboxylic acid, originating from the hydrolysis of the ester in the reaction medium, into acyl chloride. Simultaneously, dehydrating agent PPA aids in the removal of water molecules from the reaction mixture. In the second approach, enamine 18 was generated under neat conditions for 2.5 h at 110 °C, followed by the addition of diphenyl ether and heating to 228–232 °C for 1.5 h. Unfortunately, specific yields were not provided in this case [1][15][16].
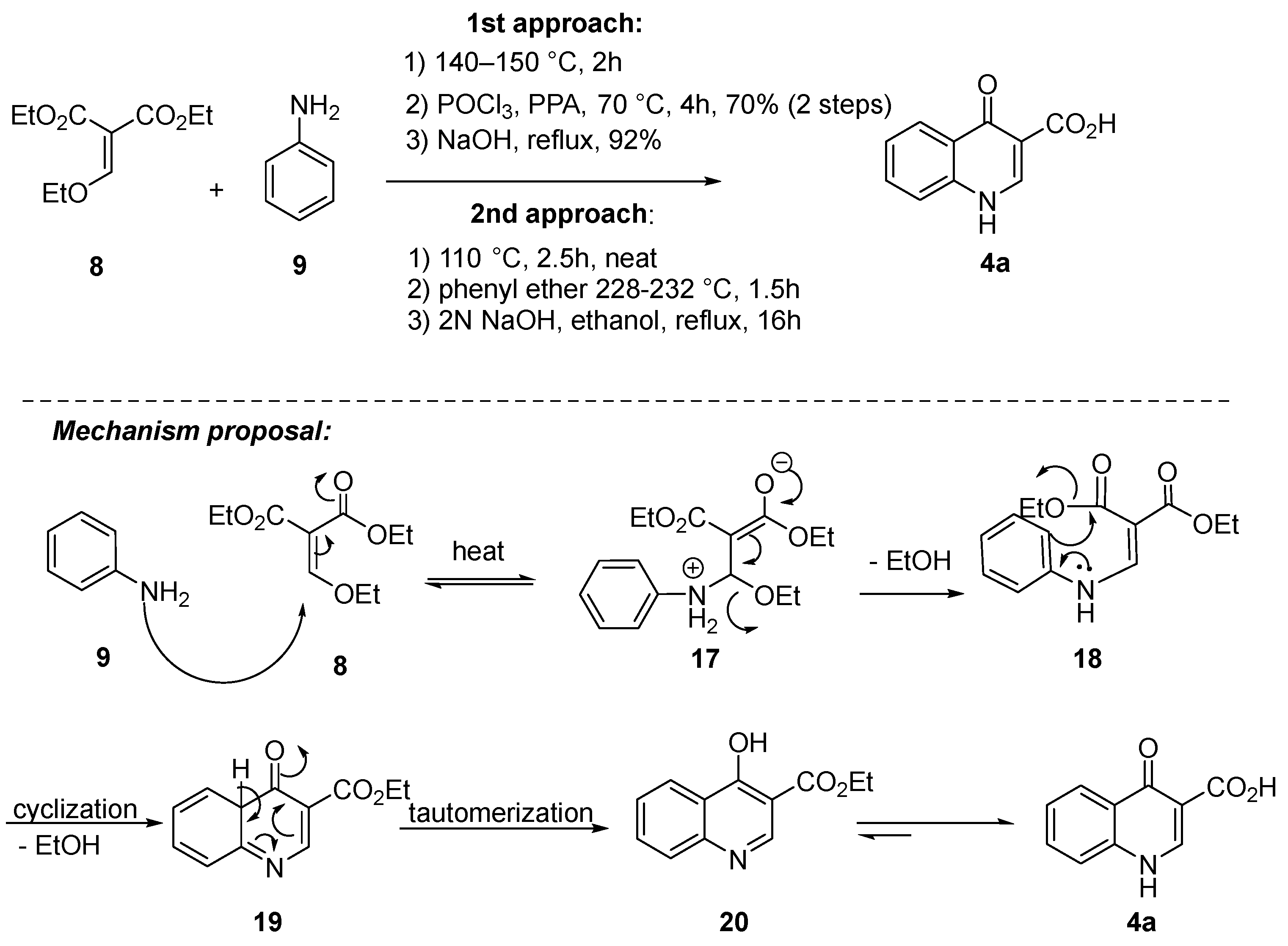
Scheme 4. Gould–Jacobs reaction as a key step for the synthesis of quinoline 4a.
Since the coupling step with quinoline carboxylic acid 4a may occur with aniline 5a or 5b, two reports were described for them. In the first report, a four-step route involved the protection of di-tert-butylphenol 21 to produce the methyl carbonate derivative 22 followed by nitration of 22 to afford 23 as a mixture of 8:1 of the desired 5-nitro regioisomer 23 and the undesired 6-nitro regioisomer. After hydrolysis with KOH in MeOH and purification by column chromatography, nitrophenol 24 was isolated in a 29% yield. Reduction of the nitro group employing transfer hydrogenation with ammonium formate led to 5a in quantitative yield. In the second approach, nitration of carbonate 22 was performed in dichloromethane at −5 to 0 °C, and carbonate 23 was isolated by crystallization from hexane without the need for chromatography. The step of reduction of the nitro group occurred without deprotection of 23 by Pd catalyzed hydrogenation with a 2 bar hydrogen gas in MeOH, and product 5b was purified by crystallization from MeOH/water (Scheme 5) [15][16]. The electron-withdrawing carbonate group may allow for nitration to occur primarily ortho/para to the tert-butyl groups since it minimizes the ortho/para-directing effect of the oxygen substituent. Data suggest that the second approach is the manufacturing of one European Public Assessment Report: Symkevi® (26 July 2018) [17].
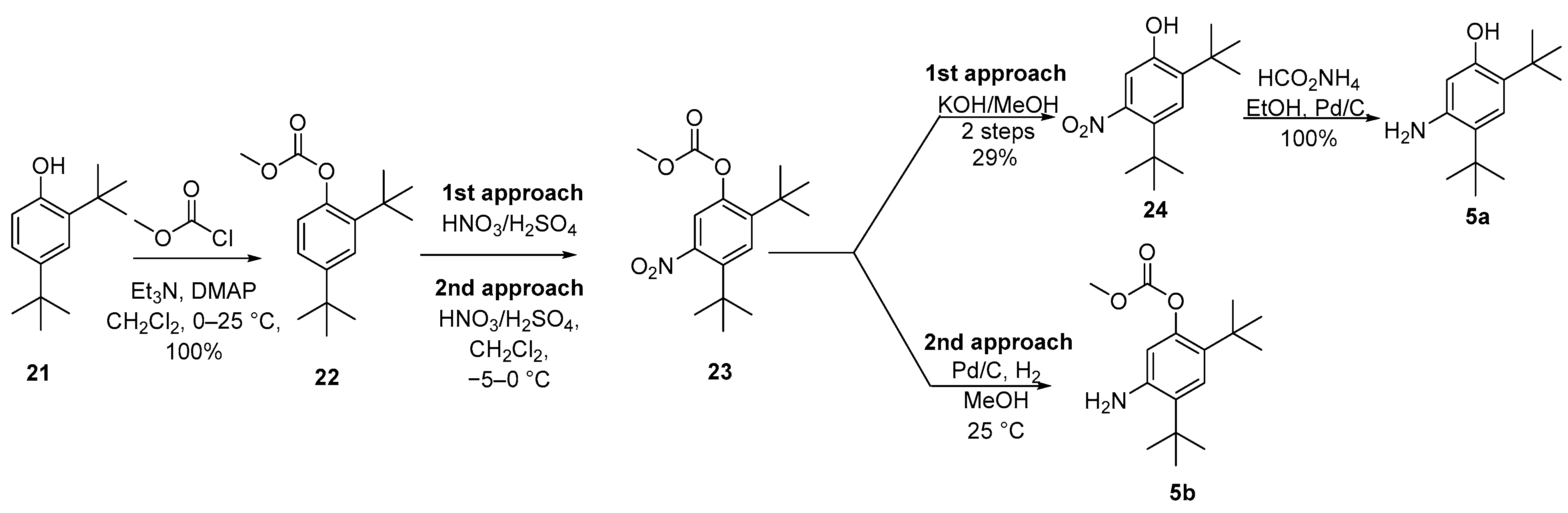
Scheme 5. Synthesis of 5-amino-2,4-di-tert-butylphenol (5a) and 5-amino-2,4-di-tert-butylphenyl methyl carbonate (5b).
2. Mechanisms of Action
Although the identification of ivacaftor by high-throughput (HT) screenings occurred in 2006, the first publications refer to 2009 as its discovery date. While ivacaftor was still in clinical development, Van Goor and colleagues [18] described the initial pharmacological properties of this potentiator in vitro, reporting its ability to partially restore CFTR activity. In this study, ivacaftor was found to increase the CFTR-mediated transepithelial current by increasing the CFTR Po, specifically after its activation by protein kinase A (PKA), in both cell lines and primary bronchial epithelial cells carrying variants p.PheF508del and/or p.Gly551Asp (legacy name G551D). The authors suggested that ivacaftor likely increases CFTR gating activity by directly binding to the protein [18], although at that time it was not yet elucidated whether it could instead act on an associated kinase or phosphatase.
The clinical approval of ivacaftor by both the US Food and Drug Administration (FDA) and the European Medicine Agency (EMA) came in 2012 initially for PwCF carrying at least one p.Gly551Asp [19] and extended in the following years for several gating/conductance variants [20][21][22]. In the same year of its approval, Eckford and collaborators [23] provided further insight into the MoA of ivacaftor by using a reconstitution system for purified CFTR protein. In this study, ivacaftor was found to bind directly to wild-type (WT) and mutant CFTR and stimulate the channel activity in an ATP-independent PKA phosphorylation-dependent process. This evidence suggested that ivacaftor binds to an allosteric site, distinct from the canonical, catalytic site, thus mediating CFTR channel potentiation by a nonconventional, ATP-independent mechanism of gating [23]. In a subsequent study, Jih and Hwang [24] performed electrophysiological recordings and used these results to propose a unified theory for the MoA of ivacaftor. Based on a CFTR gating model, in the presence of ATP, ATP-independent gating is an inherent component of the gating transitions observed for WT-CFTR. Additionally, this model includes a flexible coupling between ATP hydrolysis and gating events. These findings indicated that ivacaftor potentiates CFTR by promoting decoupling between gating cycles and ATP hydrolysis. The authors also suggested that the binding site of ivacaftor in CFTR is unlikely to be on the RD but instead may be located on the TMDs [24].
Two publications from independent research groups investigated whether ivacaftor was involved in the limited improvement of p.Phe508del-CFTR when co-administered with correctors, namely lumacaftor and tezacaftor [25][26]. Cholon and colleagues observed that, in primary cells, chronic exposure to ivacaftor promotes inhibition of lumacaftor-rescued p.Phe508del-CFTR by decreasing the stability of the corrected protein in a dose-dependent manner [25]. In parallel, Veit and collaborators also demonstrated that exposure to ivacaftor decreases the rescue efficacy of lumacaftor and tezacaftor in p.Phe508del-CFTR-expressing immortalized and primary cells [26]. Prolonged exposure (24 h) to ivacaftor reduced not only the folding efficiency but also the biochemical stability of p.Phe508del-CFTR. These findings were further confirmed by subsequent publications, demonstrating an inhibitory effect of ivacaftor on lumacaftor- or tezacaftor-rescued p.Phe508del-CFTR PM expression [27]. Similar effects were also observed in a later study in which long-term exposure to ivacaftor decreased elexacaftor-rescued p.Phe508del-CFTR [28]. In dynamics simulation and molecular docking studies, a putative binding site was proposed for ivacaftor in a region localized in the NBD1:NBD2 interface and the coupling helix of the intracellular loop (ICL) 1 [26].
In 2015, Yeh and co-authors [29] delved into the understanding of the modulation of CFTR channel gating by permeant ions, namely nitrate and ivacaftor. In this study, similarly to ivacaftor, nitrate increased the Po of the WT-CFTR channel, indicating that it has potentiator activity. Additive effects on CFTR gating were observed when both molecules were used together, suggesting that they work independently, through different binding sites to stimulate CFTR activity. Furthermore, this study proposed a putative binding site for ivacaftor on the TMDs of CFTR, at the interface between the membrane lipids and the protein channel [29]. These authors performed subsequent studies using electrophysiological recordings to further investigate ivacaftor and ABBV-974 (formerly GLPG1837) [30]. Interestingly, both potentiators were found to share a common mechanism to stimulate CFTR gating by competing for the same binding site, notwithstanding their variations in chemical structure, affinity, potency, and efficiency. This study also served as a basis to propose a four-state kinetic model based on a classic allosteric modulation model to explain the MoA and energetic coupling of potentiator binding and the opening of the CFTR channel [30]. In the following assessments, in silico molecular docking and electrophysiological assays were combined to provide further evidence of binding sites for ivacaftor and ABBV-974 [31]. Data from this study allowed for the identification of two potential binding sites located at the TMD1/2 interface, reinforcing the previous findings indicating that ivacaftor and ABBV-974 share the same binding site.
Another putative binding site for ivacaftor was proposed in the study of Byrnes and colleagues [32] using hydrogen/deuterium exchange (HDX) mass spectrometry to characterize CFTR conformational dynamics and its binding interactions with ligands. Using this approach, ivacaftor was suggested to bind to a region of amino acids in ICL4 of the “ball and socket” joint at the TMD2:NBD1 interface—a region close to where the p.Phe508 residue is present. This implies that HDX protection by ivacaftor at ICL4 may extend to the region of this residue as well. Using another approach, Csanády and Töröcsik [33] explored the solubility profile and potency of ivacaftor by stimulating WT and mutant CFTR channels in cell-free membrane patches. It was found that the aqueous solubility of ivacaftor is two orders of magnitude lesser than previously suggested [34]. Furthermore, CFTR stimulation by ivacaftor in cell-free patches was shown to be fully reversible, contrary to what was previously reported [24][29][30][35][36][37]. Based on these observations, the authors mentioned that up to that point, ivacaftor effects were assessed only at high supersaturated concentrations. This study also proposed a kinetic model for the MoA of ivacaftor in CFTR potentiation consisting of two independent binding sites with identical affinity, which thus requires that two molecules bind to the channel to potentiate CFTR [33].
Using cryo-EM, Liu and collaborators [38] were able to determine the structure of CFTR in complex with ivacaftor (PDB: 6O2P; Figure 1) and separately with ABBV-974 (PDB: 6O1V), allowing for more direct evidence of these potentiators’ binding site. These results validated previous reports that suggested a common binding site for both ivacaftor and ABBV-974 located in a region inside the lipid bilayer, within the protein–lipid interface of TMD1/2. This hotspot coincides with a hinge region involved in the gating of the channel. Such evidence allowed the authors to suggest that the open configuration of the CFTR channel is stabilized (instead of the closed) when a drug is present in that binding pocket [38]. A subsequent study by Righetti and collaborators [39] used this recently determined structure of human CFTR in complex with ivacaftor [38] to investigate the binding of potentiator to p.Phe508del-CFTR by combining molecular docking, pharmacophore mapping, and quantitative structure–activity relationship (SAR) analysis. By exploiting the most relevant amino acids involved in the ivacaftor binding site, the authors found key residues such as p.Phe931 and p.Arg933 that act via Van der Waals interactions, H-bonds, cation-π contacts and involve additional polar interactions in other residues to stabilize the binding of potentiators like ivacaftor [39].
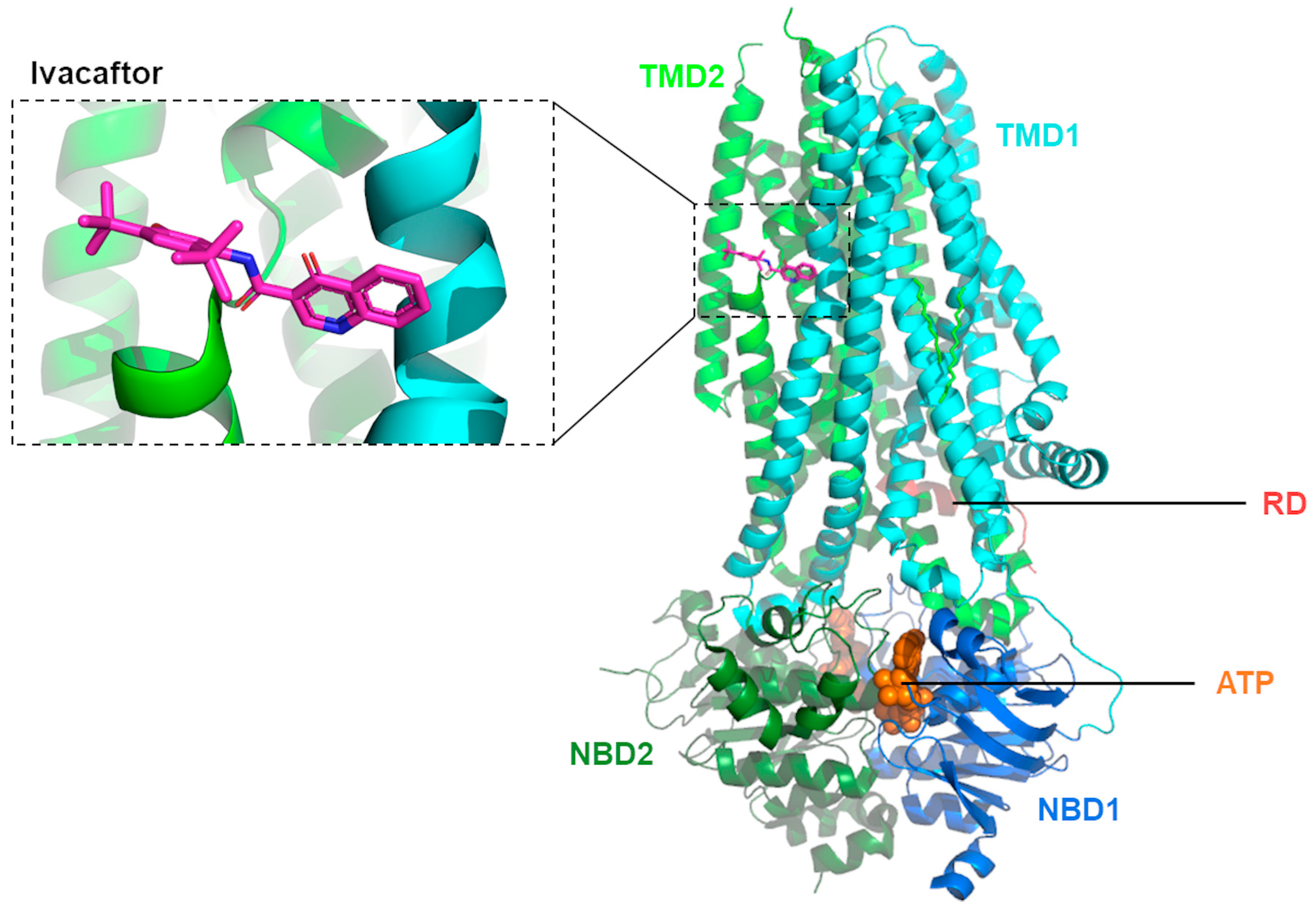
Figure 1. Molecular structure of phosphorylated, ATP-bound human CFTR in complex with ivacaftor. Ribbon diagram adapted from PDB: 6O2P [38] using PyMol Version 2.5.7.
Observing such contradictory results regarding the MoA of CFTR potentiation by ivacaftor, Laselva and collaborators [40] decided to re-evaluate these putative binding sites in the natural context of this channel protein in the lipid bilayer by using photoactivatable ivacaftor probe analogs. With the evidence obtained in this study, the authors proposed a model with two specific binding sites for ivacaftor: one in ICL4 at the NBD1:TMD2 interface and the other in the region of TMD1/2 and membrane lipid interface, as identified by cryo-EM [38]. Whether ivacaftor stabilizes CFTR open channel configuration by binding to ICL4 independently or in conjunction with its binding in the other region remains to be further elucidated [40].
Recently, Levring and collaborators [41] explored SAR in human WT-CFTR at a single-molecule resolution, combining ensemble ATPase activity measurements, single-molecule fluorescence resonance energy transfer (FRET), imaging, electrophysiology, and kinetic simulations. Using this integrative approach, the authors showed the occurrence of dimerization of the two NBDs before the channel opening, revealing an allosteric gating mechanism involving the channel pore and the catalytical binding site. Furthermore, it was observed that potentiators ivacaftor and ABBV-974 act on CFTR and enhance channel activity by increasing pore opening while the NBDs are dimerized, thus influencing the coupling efficiency between ion permeation and NBD dimerization [41]. More recent insights into the mechanisms underlying the action of ivacaftor were described by Ersoy and colleagues [42] who investigated allosteric communications in the CFTR protein by using computational analysis. The authors observed that the binding site for ivacaftor comprises some residues that are main allosteric sources, suggesting a role for this compound as an allosteric modulator. Furthermore, it was found that ivacaftor’s binding site shares similarities with the ATP binding site as both send information to an almost identical set of residues, suggesting that ivacaftor indirectly increases the Po by replicating the combined allosteric signaling triggered by the binding of ATP and by gating residues [42].
It should be noted that, despite the therapeutic accomplishments of ivacaftor, it only attains a partial restoration of the CFTR gating activity [18][24][43]. Accordingly, novel potentiators have been investigated, and their combination with complementary mechanisms (i.e., co-potentiators) has emerged as a strategy to further enhance CFTR gating activity [43][44][45][46][47][48].
References
- Hughes, D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322.
- Gould, R.G.; Jacobs, W.A. The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895.
- Zewge, D.; Chen, C.-Y.; Deer, C.; Dormer, P.G.; Hughes, D.L. A Mild and Efficient Synthesis of 4-Quinolones and Quinolone Heterocycles. J. Org. Chem. 2007, 72, 4276–4279.
- Desi Reddy, S.R.; Rane, D.R.; Velivela, V.S.R. Industrial Process for Making Ivacaftor and Its Intermediates. U.S. Patent 2017/0096397 A1, 6 April 2017.
- Zhang, R.; Han, G.; Jiang, L.; Shen, Y.; Yang, R.; Mao, Y.; Wang, H. An Efficient Synthesis of Ivacaftor. J. Heterocycl. Chem. 2017, 54, 3169–3173.
- Thatipally, S.; Reddy, V.K.; Dammalapati, V.L.N.R.; Chava, S. Processes for the Preparation of Ivacaftor. U.S. Patent 2018/0244622 A1, 30 August 2018.
- Vasudevan, N.; Jachak, G.R.; Reddy, D.S. Breaking and Making of Rings: A Method for the Preparation of 4-Quinolone-3-carb oxylic Acid Amides and the Expensive Drug Ivacaftor. European J. Org. Chem. 2015, 2015, 7433–7437.
- Reddy, D.S.; Kulkarni, A.A.; Natarajan, V.; Sharma, M.K. An Improved Process for the Synthesis of Ivacaftor. Europe Patent 3464243 A2, 10 April 2019.
- Thatipally, S.; Dammalapati, V.L.N.R.; Gorantla, S.R.; Chava, S. A Process for the Preparation of Ivacaftor and Its Intermediates. U.S. Patent WO 2014/125506 A2, 21 August 2014.
- Hadida, S.; Van Goor, F.; Zhou, J.; Arumugam, V.; McCartney, J.; Hazlewood, A.; Decker, C.; Negulescu, P.; Grootenhuis, P.D.J. Discovery of N-(2,4-Di-Tert-Butyl-5-Hydroxyphenyl)-4-Oxo-1,4-Dihydroquinoline-3-Carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. J. Med. Chem. 2014, 57, 9776–9795.
- Hadida-Ruah, S.; Hazelwood, A.; Van Goor, F.; Singh, A.; Zhou, J.; McCartney, J. Modulators of ATP-Binding Cassette Transporters. U.S. Patent 2006/0074075 A1, 6 April 2006.
- Hadida-Ruah, S.; Hazelwood, A.; Grootenhuis, P.; Van Goor, F.; Singh, A.; Zhou, J.; McCartney, J. Modulators of ATP-Binding Cassette Transporters. U.S. Patent 7,495,103 B2, 24 February 2009.
- Lai, C.K.; Song, T.V.; Li, J.J. Ivacaftor (Kalydeco): A CFTR Potentiator for the Treatment of Cystic Fibrosis. In Innovative Drug Synthesis; Li, J.J., Johnson, D.S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 303–316. ISBN 9781118819951.
- Li, J.J. Name Reactions, 3rd ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-30030-4.
- Arekar, S.G.; Johnston, S.C.; Krawiec, M.; Medek, A.; Mudunuri, P.; Sullivan, M.J. Solid Forms of N--1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. U.S. Patent 2011/0230519 A1, 22 September 2011.
- Luisi, B.; Arekar, S.G.; Costache, A.; Dinehart, K.R.; Johnston, S.C.; Neubert-Langille, B.J. Solid Forms of N--1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. U.S. Patent 9,045,425 B2, 2 June 2015.
- European Medicines Agency. European Public Assessment Report: Symkevi (International Non-Proprietary Name: Tezacaftor/Ivacaftor). Procedure No. EMEA/H/C/004682/0000. 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/symkevi-epar-public-assessment-report_en.pdf (accessed on 22 December 2023).
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF Airway Epithelial Cell Function in Vitro by a CFTR Potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830.
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672.
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR Modulators for the Treatment of Cystic Fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913.
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis and a Non-G551D Gating Mutation. J. Cyst. Fibros. 2014, 13, 674–680.
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M.; GOALe(2) Investigators. Effectiveness of Ivacaftor in Cystic Fibrosis Patients with Non-G551D Gating Mutations. J. Cyst. Fibros. 2019, 18, 102–109.
- Eckford, P.D.W.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-Dependent but ATP-Independent Manner. J. Biol. Chem. 2012, 287, 36639–36649.
- Jih, K.-Y.; Hwang, T.-C. Vx-770 Potentiates CFTR Function by Promoting Decoupling between the Gating Cycle and ATP Hydrolysis Cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409.
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator Ivacaftor Abrogates Pharmacological Correction of ΔF508 CFTR in Cystic Fibrosis. Sci. Transl. Med. 2014, 6, 246ra96.
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.-W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.-S.; Finkbeiner, W.E.; et al. Some Gating Potentiators, Including VX-770, Diminish ΔF508-CFTR Functional Expression. Sci. Transl. Med. 2014, 6, 246ra97.
- Lopes-Pacheco, M.; Silva, I.A.L.; Turner, M.J.; Carlile, G.W.; Sondo, E.; Thomas, D.Y.; Pedemonte, N.; Hanrahan, J.W.; Amaral, M.D. Characterization of the Mechanism of Action of RDR01752, a Novel Corrector of F508del-CFTR. Biochem. Pharmacol. 2020, 180, 114133.
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric Folding Correction of F508del and Rare CFTR Mutants by Elexacaftor-Tezacaftor-Ivacaftor (Trikafta) Combination. JCI Insight 2020, 5, e139983.
- Yeh, H.-I.; Yeh, J.-T.; Hwang, T.-C. Modulation of CFTR Gating by Permeant Ions. J. Gen. Physiol. 2015, 145, 47–60.
- Yeh, H.-I.; Sohma, Y.; Conrath, K.; Hwang, T.-C. A Common Mechanism for CFTR Potentiators. J. Gen. Physiol. 2017, 149, 1105–1118.
- Yeh, H.-I.; Qiu, L.; Sohma, Y.; Conrath, K.; Zou, X.; Hwang, T.-C. Identifying the Molecular Target Sites for CFTR Potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019, 151, 912–928.
- Byrnes, L.J.; Xu, Y.; Qiu, X.; Hall, J.D.; West, G.M. Sites Associated with Kalydeco Binding on Human Cystic Fibrosis Transmembrane Conductance Regulator Revealed by Hydrogen/Deuterium Exchange. Sci. Rep. 2018, 8, 4664.
- Csanády, L.; Töröcsik, B. Cystic Fibrosis Drug Ivacaftor Stimulates CFTR Channels at Picomolar Concentrations. Elife 2019, 8, e46450.
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of Ivacaftor on CFTR Forms with Missense Mutations Associated with Defects in Protein Processing or Function. J. Cyst. Fibros. 2014, 13, 29–36.
- Lin, W.-Y.; Sohma, Y.; Hwang, T.-C. Synergistic Potentiation of Cystic Fibrosis Transmembrane Conductance Regulator Gating by Two Chemically Distinct Potentiators, Ivacaftor (VX-770) and 5-Nitro-2-(3-Phenylpropylamino) Benzoate. Mol. Pharmacol. 2016, 90, 275–285.
- Wang, Y.; Liu, J.; Loizidou, A.; Bugeja, L.A.; Warner, R.; Hawley, B.R.; Cai, Z.; Toye, A.M.; Sheppard, D.N.; Li, H. CFTR Potentiators Partially Restore Channel Function to A561E-CFTR, a Cystic Fibrosis Mutant with a Similar Mechanism of Dysfunction as F508del-CFTR. Br. J. Pharmacol. 2014, 171, 4490–4503.
- Wang, Y.; Cai, Z.; Gosling, M.; Sheppard, D.N. Potentiation of the Cystic Fibrosis Transmembrane Conductance Regulator Cl− Channel by Ivacaftor Is Temperature Independent. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L846–L857.
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural Identification of a Hotspot on CFTR for Potentiation. Science 2019, 364, 1184–1188.
- Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445.
- Laselva, O.; Qureshi, Z.; Zeng, Z.-W.; Petrotchenko, E.V.; Ramjeesingh, M.; Hamilton, C.M.; Huan, L.-J.; Borchers, C.H.; Pomès, R.; Young, R.; et al. Identification of Binding Sites for Ivacaftor on the Cystic Fibrosis Transmembrane Conductance Regulator. iScience 2021, 24, 102542.
- Levring, J.; Terry, D.S.; Kilic, Z.; Fitzgerald, G.; Blanchard, S.C.; Chen, J. CFTR Function, Pathology and Pharmacology at Single-Molecule Resolution. Nature 2023, 616, 606–614.
- Ersoy, A.; Altintel, B.; Livnat Levanon, N.; Ben-Tal, N.; Haliloglu, T.; Lewinson, O. Computational Analysis of Long-Range Allosteric Communications in CFTR. Elife 2023, 12, RP88659.
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-Specific Dual Potentiators Maximize Rescue of CFTR Gating Mutants. J. Cyst. Fibros. 2020, 19, 236–244.
- Phuan, P.W.; Son, J.H.; Tan, J.A.; Li, C.; Musante, I.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Kurth, M.J.; Galietta, L.J.; et al. Combination Potentiator (‘co-Potentiator’) Therapy for CF Caused by CFTR Mutants, Including N1303K, That Are Poorly Responsive to Single Potentiators. J. Cyst. Fibros. 2018, 17, 595–606.
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor Co-Potentiates the Activity of F508del and Gating Mutants of CFTR. J. Cyst. Fibros. 2021, 20, 895–898.
- Liu, J.; Berg, A.P.; Wang, Y.; Jantarajit, W.; Sutcliffe, K.J.; Stevens, E.B.; Cao, L.; Pregel, M.J.; Sheppard, D.N. A Small Molecule CFTR Potentiator Restores ATP-dependent Channel Gating to the Cystic Fibrosis Mutant G551D-CFTR. Br. J. Pharmacol. 2022, 179, 1319–1337.
- Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Cuyx, S.; Van Biervliet, S.; Dupont, L.; Christ, F.; Debyser, Z.; Vermeulen, F.; Carlon, M.S. Novel CFTR Modulator Combinations Maximise Rescue of G85E and N1303K in Rectal Organoids. ERJ Open Res. 2022, 8, 00716–02021.
- Bacalhau, M.; Ferreira, F.C.; Silva, I.A.L.; Buarque, C.D.; Amaral, M.D.; Lopes-Pacheco, M. Additive Potentiation of R334W-CFTR Function by Novel Small Molecules. J. Pers. Med. 2023, 13, 102.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Organic Synthesis
Revisions:
2 times
(View History)
Update Date:
17 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No