 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Allison B. Reiss | -- | 3126 | 2024-02-09 02:09:19 | | | |
| 2 | Lindsay Dong | Meta information modification | 3126 | 2024-02-11 12:35:03 | | |
Video Upload Options
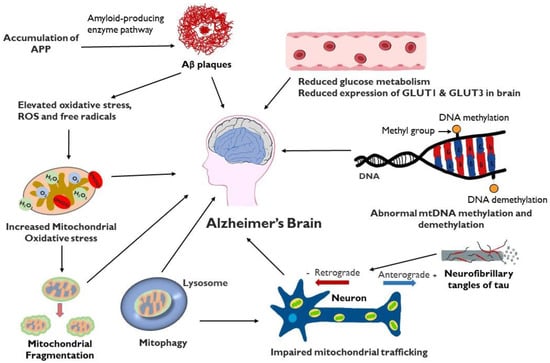
Alzheimer’s disease (AD) is a progressive and incurable neurodegenerative disorder that primarily affects persons aged 65 years and above. It causes dementia with memory loss and deterioration in thinking and language skills. AD is characterized by specific pathology resulting from the accumulation in the brain of extracellular plaques of amyloid-β and intracellular tangles of phosphorylated tau. The importance of mitochondrial dysfunction in AD pathogenesis, while previously underrecognized, is now more and more appreciated. Mitochondria are an essential organelle involved in cellular bioenergetics and signaling pathways. Mitochondrial processes crucial for synaptic activity such as mitophagy, mitochondrial trafficking, mitochondrial fission, and mitochondrial fusion are dysregulated in the AD brain. Excess fission and fragmentation yield mitochondria with low energy production. Reduced glucose metabolism is also observed in the AD brain with a hypometabolic state, particularly in the temporo-parietal brain regions.
1. Introduction
2. Mitochondrial ATP Production and Oxidative Stress in Neurons
2.1. Structural Characteristics
2.2. Energy Production by Mitochondria and Mitochondrial Oxidative Stress
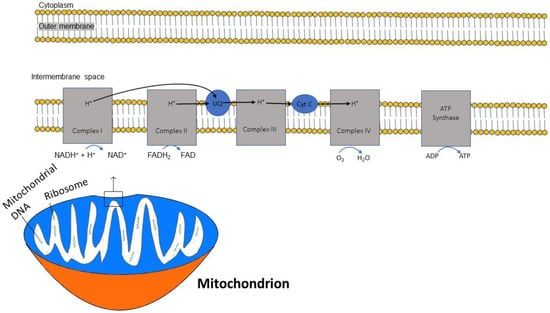
2.3. ATP and Oxidative Phosphorylation
3. Mitochondrial Trafficking
4. Mitophagy, Mitochondrial Dynamics and AD
4.1. Mitophagy
4.2. Aβ and Tau in Mitophagy and Mitochondrial Movement
4.3. Mitochondrial Fission and Fusion
4.4. Effects of Amyloid and Tau on Fission and Fusion
5. Mitochondrial DNA Methylation
6. Glucose Metabolism Reduced in AD
7. Apolipoprotein (Apo)E Gene Impact on Mitochondria and Bioenergetics
8. Mitochondria in the Treatment of AD
9 Conclusions
References
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265.
- Volloch, V.; Rits-Volloch, S. Effect of Lecanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. J. Alzheimers Dis. 2023, 93, 1277–1284.
- Varadharajan, A.; Davis, A.D.; Ghosh, A.; Jagtap, T.; Xavier, A.; Menon, A.J.; Roy, D.; Gandhi, S.; Gregor, T. Guidelines for pharmacotherapy in Alzheimer’s disease—A primer on FDA-approved drugs. J. Neurosci. Rural Pract. 2023, 14, 566–573.
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell. Res. 2018, 27, 121–130.
- Tapias, V.; González-Andrés, P.; Peña, L.F.; Barbero, A.; Núñez, L.; Villalobos, C. Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease. Antioxidants 2023, 12, 1282.
- Dentoni, G.; Castro-Aldrete, L.; Naia, L.; Ankarcrona, M. The Potential of Small Molecules to Modulate the Mitochondria-Endoplasmic Reticulum Interplay in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 920228.
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604.
- Bonda, D.J.; Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial dynamics in Alzheimer’s disease: Opportunities for future treatment strategies. Drugs Aging 2010, 27, 181–192.
- Mei, T.; Li, Y.; Orduña Dolado, A.; Li, Z.; Andersson, R.; Berliocchi, L.; Rasmussen, L.J. Pooled analysis of frontal lobe transcriptomic data identifies key mitophagy gene changes in Alzheimer’s disease brain. Front. Aging Neurosci. 2023, 15, 1101216.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296.
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208.
- Atlante, A.; Valenti, D. Mitochondria Have Made a Long Evolutionary Path from Ancient Bacteria Immigrants within Eukaryotic Cells to Essential Cellular Hosts and Key Players in Human Health and Disease. Curr. Issues Mol. Biol. 2023, 45, 4451–4479.
- Joshi, A.; Richard, T.H.; Gohil, V.M. Mitochondrial phospholipid metabolism in health and disease. J. Cell Sci. 2023, 136, jcs260857.
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492.
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360.
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777.
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343.
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890.
- Caruana, N.J.; Stroud, D.A. The road to the structure of the mitochondrial respiratory chain supercomplex. Biochem. Soc. Trans. 2020, 48, 621–629.
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15.
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Bio. 2020, 37, 101674.
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76.
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomedicines 2023, 11, 532.
- Han, Y.; Liu, D.; Cheng, Y.; Ji, Q.; Liu, M.; Zhang, B.; Zhou, S. Maintenance of mitochondrial homeostasis for Alzheimer’s disease: Strategies and challenges. Redox Biol. 2023, 63, 102734.
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824.
- Biffi, A.; Sabuncu, M.R.; Desikan, R.S.; Schmansky, N.; Salat, D.H.; Rosand, J.; Anderson, C.D.; Alzheimer’s disease Neuroimaging Initiative (ADNI). Genetic variation of oxidative phosphorylation genes in stroke and Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1956.e1–1956.e8.
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 2015, 44, 375–378.
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport: Driving synaptic function. Science 2019, 366, eaaw9997.
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. J. Clin. Investg. 2023, 133, e168554.
- Niescier, R.F.; Kwak, S.K.; Joo, S.H.; Chang, K.T.; Min, K.T. Dynamics of Mitochondrial Transport in Axons. Front. Cell. Neurosci. 2016, 10, 123.
- Hung, C. Importance of retrograde axonal transport in mitochondrial health and distribution. Cell Death Discov. 2021, 7, 106.
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444.
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185.
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207.
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806.
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253.
- Varte, V.; Munkelwitz, J.W.; Rincon-Limas, D.E. Insights from Drosophila on Aβ- and tau-induced mitochondrial dysfunction: Mechanisms and tools. Front. Neurosci. 2023, 17, 1184080.
- Wang, Z.T.; Lu, M.H.; Zhang, Y.; Ji, W.L.; Lei, L.; Wang, W.; Fang, L.P.; Wang, L.W.; Yu, F.; Wang, J.; et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 2019, 18, e12860.
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166.
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 2498–2507.
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes, and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 147, 291–309.
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol. 2016, 26, 632–647.
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488.
- Averchuk, A.S.; Ryazanova, M.V.; Baranich, T.I.; Stavrovskaya, A.V.; Rozanova, N.A.; Novikova, S.V.; Salmina, A.B. The Neurotoxic Effect of β-Amyloid Is Accompanied by Changes in the Mitochondrial Dynamics and Autophagy in Neurons and Brain Endothelial Cells in the Experimental Model of Alzheimer’s Disease. Bull. Exp. Biol. Med. 2023, 175, 315–320.
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150.
- Dou, Y.; Tan, Y. Presequence protease reverses mitochondria-specific amyloid-β-induced mitophagy to protect mitochondria. FASEB J. 2023, 37, e22890.
- Limorenko, G.; Lashuel, H.A. Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target tauopathies. Chem. Soc. Rev. 2022, 51, 513–565.
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21.
- Saavedra, J.; Nascimento, M.; Liz, M.A.; Cardoso, I. Key brain cell interactions and contributions to the pathogenesis of Alzheimer’s disease. Front. Cell Dev. Biol. 2022, 10, 1036123.
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148.
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713.
- Eckert, A.; Schulz, K.L.; Rhein, V.; Götz, J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 2010, 41, 107–114.
- Cario, A.; Berger, C.L. Tau, microtubule dynamics, and axonal transport: New paradigms for neurodegenerative disease. Bioessays 2023, 45, e2200138.
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. Neurosci. Ther. 2011, 17, 514–524.
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95.
- Liu, X.; Ye, M.; Ma, L. The emerging role of autophagy and mitophagy in tauopathies: From pathogenesis to translational implications in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1022821.
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667.
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368.
- Lechado-Terradas, A.; Schepers, S.; Zittlau, K.I.; Sharma, K.; Ok, O.; Fitzgerald, J.C.; Geimer, S.; Westermann, B.; Macek, B.; Kahle, P.J. Parkin-dependent mitophagy occurs via proteasome-dependent steps sequentially targeting separate mitochondrial sub-compartments for autophagy. Autophagy Rep. 2022, 1, 576–602.
- Das, R.; Chakrabarti, O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020, 48, 631–644.
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 8, 1240–1247.
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509.
- Dhapola, R.; Sarma, P.; Medhi, B.; Prakash, A.; Reddy, D.H. Recent Advances in Molecular Pathways and Therapeutic Implications Targeting Mitochondrial Dysfunction for Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 535–555.
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834.
- Abtahi, S.L.; Masoudi, R.; Haddadi, M. The distinctive role of tau and amyloid beta in mitochondrial dysfunction through alteration in Mfn2 and Drp1 mRNA Levels: A comparative study in Drosophila melanogaster. Gene 2020, 754, 144854.
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946.
- Nakamura, T.; Lipton, S.A. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: Potential implications for Alzheimer’s and Parkinson’s diseases. Apoptosis 2010, 15, 1354–1363.
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal mitochondria modulation of LPS-induced neuroinflammation. J. Neurosci. 2020, 40, 1756–1765.
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J. Neuroinflammation 2021, 18, 54.
- Vijayan, M.; Reddy, P.H. Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 8561.
- Bera, A.; Lavanya, G.; Reshmi, R.; Dev, K.; Kumar, R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of Alzheimer’s disease. Eur. J. Neurosci. 2022, 9, 5516–5531.
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118664.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554.
- Amaral, A.C.; Perez-Nievas, B.G.; Siao Tick Chong, M.; Gonzalez-Martinez, A.; Argente-Escrig, H.; Rubio-Guerra, S.; Commins, C.; Muftu, S.; Eftekharzadeh, B.; Hudry, E.; et al. Isoform-selective decrease of glycogen synthase kinase-3-beta (GSK-3β) reduces synaptic tau phosphorylation, transcellular spreading, and aggregation. iScience 2021, 24, 102058.
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112.
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 2016, 4, 814–828.
- Coppedè, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 86.
- Coppedè, F. Mitochondrial DNA methylation and mitochondria-related epigenetics in neurodegeneration. Neural Regen. Res. 2024, 19, 405–406.
- Xu, Y.; Xu, L.; Han, M.; Liu, X.; Li, F.; Zhou, X.; Wang, Y.; Bi, J. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an app/ps1 transgenic mouse model of Alzheimer disease. Biochem. Biophys. Res. Commun. 2019, 520, 41–46.
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002.
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223.
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9, S16.
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597.
- Kumar, V.; Kim, S.H.; Bishayee, K. Dysfunctional Glucose Metabolism in Alzheimer’s Disease Onset and Potential Pharmacological Interventions. Int. J. Mol. Sci. 2022, 23, 9540.
- Patel, V.; Mill, J.; Okonkwo, O.C.; Salamat, S.; Li, L.; Raife, T. Global Energy Metabolism Deficit in Alzheimer Disease Brain. J. Prev. Alzheimers Dis. 2024, 11, 171–178.
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510.
- Myoraku, A.; Klein, G.; Landau, S.; Tosun, D.; Alzheimer’s Disease Neuroimaging, I. Regional uptakes from early frame amyloid PET and (18)F-FDG PET scans are comparable independent of disease state. Eur. J. Hybrid. Imaging 2022, 6, 2.
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Murray, J.; Li, Y.; Glodzik, L.; McHugh, P.; Williams, S.; Cummings, M.; Pirraglia, E.; et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol. Aging 2013, 34, 22–34.
- Blazhenets, G.; Ma, Y.; Sörensen, A.; Schiller, F.; Rücker, G.; Eidelberg, D.; Frings, L.; Meyer, P.T.; Alzheimer Disease Neuroimaging Initiative. Predictive value of 18F-florbetapir and 18F-FDG PET for conversion from mild cognitive impairment to Alzheimer dementia. J. Nucl. Med. 2020, 61, 597–603.
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822.
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimers Res. Ther. 2019, 11, 57.
- Chen, P.; Shen, Z.; Wang, Q.; Zhang, B.; Zhuang, Z.; Lin, J.; Shen, Y.; Chen, Y.; Dai, Z.; Wu, R. Reduced Cerebral Glucose Uptake in an Alzheimer’s Rat Model With Glucose-Weighted Chemical Exchange Saturation Transfer Imaging. Front. Aging Neurosci. 2021, 13, 618690.
- Zhou, Y.; Dougherty, J.H., Jr.; Hubner, K.F.; Bai, B.; Cannon, R.L.; Hutson, R.K. Abnormal connectivity in the posterior cingulate and hippocampus in early Alzheimer’s disease and mild cognitive impairment. Alzheimers Dement. 2008, 4, 265–270.
- Ferrari, B.L.; Neto, G.C.C.; Nucci, M.P.; Mamani, J.B.; Lacerda, S.S.; Felicio, A.C.; Amaro, E., Jr.; Gamarra, L.F. The accuracy of hippocampal volumetry and glucose metabolism for the diagnosis of patients with suspected Alzheimer’s disease, using automatic quantitative clinical tools. Medicine 2019, 98, e17824.
- Weise, C.M.; Chen, K.; Chen, Y.; Kuang, X.; Savage, C.R.; Reiman, E.M.; Alzheimer’s Disease Neuroimaging Initiative. Left lateralized cerebral glucose metabolism declines in amyloid-β positive persons with mild cognitive impairment. Neuroimage Clin. 2018, 20, 286–296.
- Kyrtata, N.; Emsley, H.C.A.; Sparasci, O.; Parkes, L.M.; Dickie, B.R. A Systematic Review of Glucose Transport Alterations in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 626636.
- Nguyen, Y.T.K.; Ha, H.T.T.; Nguyen, T.H.; Nguyen, L.N. The role of SLC transporters for brain health and disease. Cell. Mol. Life Sci. 2022, 79, 20.
- Patching, S.G. Glucose transporters at the blood–brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077.
- Szablewski, L. Brain Glucose Transporters: Role in Pathogenesis and Potential Targets for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8142.
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334.
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364.
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2011, 116, 1–9.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337.
- Kang, S.S.; Ebbert, M.T.W.; Baker, K.E.; Cook, C.; Wang, X.; Sens, J.P.; Kocher, J.P.; Petrucelli, L.; Fryer, J.D. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 2018, 215, 2235–2245.
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72.
- Oliver, D.M.A.; Reddy, P.H. Molecular Basis of Alzheimer’s Disease: Focus on Mitochondria. J. Alzheimers Dis. 2019, 72, S95–S116.
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Alzheimer’s Disease Genetics Consortium Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667.
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64.
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221.
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828.
- Alshial, E.E.; Abdulghaney, M.I.; Wadan, A.S.; Abdellatif, M.A.; Ramadan, N.E.; Suleiman, A.M.; Waheed, N.; Abdellatif, M.; Mohammed, H.S. Mitochondrial dysfunction and neurological disorders: A narrative review and treatment overview. Life Sci. 2023, 334, 122257.
- Wang, M.; Xuan, T.; Li, H.; An, J.; Hao, T.; Cheng, J. Protective effect of FXN overexpression on ferroptosis in L-Glu-induced SH-SY5Y cells. Acta Histochem. 2024, 126, 152135.
- Rajkumar, M.; Govindaraj, P.; Vimala, K.; Thangaraj, R.; Kannan, S. Chitosan/PLA-loaded Magnesium oxide nanocomposite to attenuate oxidative stress, neuroinflammation and neurotoxicity in rat models of Alzheimer’s disease. Metab. Brain Dis. 2023.
- Lee, D.Y.; Lee, K.M.; Um, J.H.; Kim, Y.Y.; Kim, D.H.; Yun, J. The Natural Alkaloid Palmatine Selectively Induces Mitophagy and Restores Mitochondrial Function in an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2023, 24, 16542.
- Jia, Y.L.; Wang, W.; Han, N.; Sun, H.L.; Dong, F.M.; Song, Y.X.; Feng, R.F.; Wang, J.H. The mitochondria-targeted small molecule SS31 delays progression of behavioral deficits by attenuating β-amyloid plaque formation and mitochondrial/synaptic deterioration in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2023, 658, 36–43.
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718.
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401.
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715.
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid. Redox Signal. 2017, 27, 1048–1066.
- Mahmood, A.; Bisoyi, P.; Banerjee, R.; Yousuf, M.; Goswami, S.K. Mitoapocynin, a mitochondria targeted derivative of apocynin induces mitochondrial ROS generation and apoptosis in multiple cell types including cardiac myoblasts: A potential constraint to its therapeutic use. Mol. Cell. Biochem. 2021, 476, 2047–2059.
- Liu, N.; Lyu, X.; Zhang, X.; Zhang, F.; Chen, Y.; Li, G. Astaxanthin attenuates cognitive deficits in Alzheimer’s disease models by reducing oxidative stress via the SIRT1/PGC-1α signaling pathway. Cell Biosci. 2023, 13, 173.
- Oliyaei, N.; Moosavi-Nasab, M.; Tanideh, N.; Iraji, A. Multiple roles of fucoxanthin and astaxanthin against Alzheimer’s disease: Their pharmacological potential and therapeutic insights. Brain Res. Bull. 2023, 193, 11–21.
- Nishida, Y.; Nawaz, A.; Hecht, K.; Tobe, K. Astaxanthin as a Novel Mitochondrial Regulator: A New Aspect of Carotenoids, beyond Antioxidants. Nutrients 2021, 14, 107.
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547.
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid1-42 memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91–101.
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110.
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204.
- Sbai, O.; Bazzani, V.; Tapaswi, S.; McHale, J.; Vascotto, C.; Perrone, L. Is Drp1 a link between mitochondrial dysfunction and inflammation in Alzheimer’s disease? Front. Mol. Neurosci. 2023, 16, 1166879.
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6.
- Bhatti, J.S.; Kaur, S.; Mishra, J.; Dibbanti, H.; Singh, A.; Reddy, A.P.; Bhatti, G.K.; Reddy, P.H. Targeting dynamin-related protein-1 as a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA)–Mol. Basis Dis. 2023, 1869, 166798.
- Liu, X.; Song, L.; Yu, J.; Huang, F.; Li, Y.; Ma, C. Mdivi-1: A promising drug and its underlying mechanisms in the treatment of neurodegenerative diseases. Histol. Histopathol. 2022, 37, 505–512.
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 719.
- Vijayan, M.; Bose, C.; Reddy, P.H. Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29.
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Aβ and inhibits abnormal interactions between Aβ and Drp1 and protects Alzheimer’s disease neurons from Aβ- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395.
- Mi, Y.; Qi, G.; Brinton, R.D.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid. Redox. Signal. 2021, 34, 611–630.
- Misrani, A.; Tabassum, S.; Huo, Q.; Tabassum, S.; Jiang, J.; Ahmed, A.; Chen, X.; Zhou, J.; Zhang, J.; Liu, S.; et al. Mitochondrial Deficits With Neural and Social Damage in Early Stage Alzheimer’s Disease Model Mice. Front. Aging Neurosci. 2021, 13, 748388.


