Alzheimer’s disease (AD) is a progressive and incurable neurodegenerative disorder that primarily affects persons aged 65 years and above. It causes dementia with memory loss and deterioration in thinking and language skills. AD is characterized by specific pathology resulting from the accumulation in the brain of extracellular plaques of amyloid-β and intracellular tangles of phosphorylated tau. The importance of mitochondrial dysfunction in AD pathogenesis, while previously underrecognized, is now more and more appreciated. Mitochondria are an essential organelle involved in cellular bioenergetics and signaling pathways. Mitochondrial processes crucial for synaptic activity such as mitophagy, mitochondrial trafficking, mitochondrial fission, and mitochondrial fusion are dysregulated in the AD brain. Excess fission and fragmentation yield mitochondria with low energy production. Reduced glucose metabolism is also observed in the AD brain with a hypometabolic state, particularly in the temporo-parietal brain regions.
1. Introduction
Alzheimer’s disease (AD) manifests as progressive cognitive decline eventually ending in death. The disease-defining pathological features observed in the brain are the accumulation of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein [
1]. The mainstay FDA-approved drugs for AD treatment offer some symptomatic relief while newer immunotherapies directed against Aβ may slow the rate of cognitive decline modestly [
2,
3]. Aduhelm, one of the new anti-amyloid immunotherapies, is being discontinued by the company Biogen as part of a reprioritizing strategy
[1]. There is no cure for AD and since approaches targeting Aβ and tau have shown that these misfolded proteins are likely not causative, attention has shifted to other mechanisms, including those involving mitochondria [
4]. Mitochondria are being explored because abnormalities in this organelle are found early in the course of the disease and can lead to many of the neuron-destroying consequences of AD [
5,
6,
7]. The disruption of mitochondrial dynamics leads to mitochondrial fragmentation, generation of reactive oxygen species (ROS) and poor energy production [
8]. Defective mitophagy further aggravates this problem, which impedes the ability of the cell to dispose of the damaged mitochondria [
9].
2. Mitochondrial ATP Production and Oxidative Stress in Neurons
2.1. Structural Characteristics
Mitochondria are an essential organelle located in the cytoplasm of eukaryotic cells [
12]. They are involved in cellular bioenergetic and signaling pathways and metabolic adaptations to keep the cell and organism alive [
13]. They are vital for ATP production through oxidative phosphorylation and for maintaining calcium homeostasis [
14,
15]. Mitochondria are rod-shaped double-membrane structures ranging in length from 0.5 µm to 1 µm [
16]. The outer and inner membranes create two compartments: an intermembrane space and an inner membrane space. The inner membrane has numerous folds called cristae that serve to increase surface area and embedded within the cristae are the proteins needed for oxidative phosphorylation and ATP generation. Enclosed in the inner membrane is a mitochondrial matrix that contains the mitochondrial DNA and holds the enzymes of the citric acid cycle and fatty acid degradation.
2.2. Energy Production by Mitochondria and Mitochondrial Oxidative Stress
Mitochondria are particularly important in neurons where energy needs are disproportionately high. Neurons use 70–80% of total energy among brain cells, while glial cells use the remainder [
17]. Mitochondria supply 93% of ATP at synapses with glycolysis providing only 7% [
18,
19].
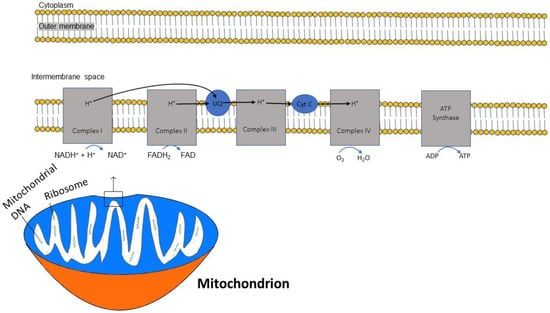
In sequential order, five multiprotein complexes (complex I, complex II, coenzyme Q, complex III, cytochrome C, and complex IV) form the electron transport chain (ETC), a chain that creates an electrochemical gradient and releases energy [
20]. (
Figure 1). The human mitochondrial genome is circularly organized and consists of 13 proteins, 22 transfer RNAs and 2 ribosomal RNAs encoded by 37 genes [
21]. Key protein subunits of complexes I–IV of the ETC are encoded by mitochondrial DNA while other subunits are encoded by nuclear DNA [
22].
Figure 1. Diagram demonstrating the flow of electrons through the mitochondrial electron transport chain (ETC). Electrons initially enter the ETC by NADH at complex I, and FADH2 from complex II. Ubiquinone transports the electrons to complex III, and then through cytochrome (Cyt) C to complex IV where oxygen is reduced into water. A proton gradient pumped across the inner mitochondrial membrane caused by the translocation of protons synthesizes ATP.
The ETC is embedded within the inner membrane of the mitochondria. ATP synthesis through the ETC is driven by the reduced form of nicotinamide adenine dinucleotide (NADH), which is generated from the citric acid cycle and serves as a donor of electrons to complex I. Two electrons from NADH are transferred to ubiquinone [
23]. This electron transfer induces the pumping of protons by complex I from the matrix to the intermembrane space, contributing to the membrane potential and energy storage for ATP production. A second entry point for electrons into the ETC is through complex II, where succinate from the citric acid cycle, when oxidized to fumarate, donates 2 electrons to the oxidized form of flavin adenine dinucleotide (FAD) in complex II to generate the reduced form FADH2. Complex II is not a proton pump and does not translocate protons and, consequently, an FADH2 molecule yields less ATP than an NADH molecule. Both complex I and complex II pass electrons to coenzyme Q at the inner mitochondrial membrane and coenzyme Q accepts electrons in pairs, transfers them to complex III, and then to cytochrome c. Once cytochrome c is reduced, it transfers electrons to complex IV (cytochrome c oxidase), where molecular oxygen (O
2) is reduced to H
2O. Lastly, complex V is a multi-subunit complex that functions under a rotational motor mechanism to allow for ATP production [
24,
25].
2.3. ATP and Oxidative Phosphorylation
There is high demand for ATP in the very metabolically active neurons in the brain and oxidative phosphorylation, occurring in the inner mitochondrial membrane, is the process in which ATP production and ROS generation are linked [
31]. Oxidative phosphorylation has been shown to play a role in AD progression, likely through oxidative damage [
32,
33,
34]. Biffi et al. used the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database to gather SNP genotype and baseline MRI results from 740 subjects in four clinical categories (cognitively normal controls, MCI non-converters, MCI converted to AD, AD) [
35]. Their analysis found that 105 genes involved in oxidative phosphorylation contributed to clinical manifestations of AD, with a major role for complex I above other complexes.
A decline in ATP is noted with oxidative stress in AD neuropathology. Oxidative stress is an early and prominent feature of AD caused by the overproduction and accumulation of ROS, which damages cells. Zhang et al. determined ATP levels in the brains of AD transgenic mice using an ATP bioluminescence assay and found that ATP content in the AD mouse brain was significantly reduced compared to wild-type C57BL/6 mice, suggesting mitochondrial dysfunction [
37].
3. Mitochondrial Trafficking
Neurons are highly polarized cells that transfer information through a combination of chemical and electrical signals at the synapse, which is distant from the cell body and maintained by axonal transport [
55,
56]. Mitochondrial trafficking in neurons is a phenomenon in which mitochondria move bidirectionally, with anterograde transport of mitochondria from the cell body to synaptic terminals and retrograde transport of mitochondria from the synaptic terminals to the cell body [
57]. Kinesin-1 mediates anterograde transport and cytoplasmic dynein motors monitor retrograde mitochondrial transport [
58].
4. Mitophagy, Mitochondrial Dynamics and AD
4.1. Mitophagy
Mitophagy, which is a mode of autophagy specifically for mitochondria, is the process of selectively degrading damaged or unneeded mitochondria, which is essential for mitochondrial quality control [
61,
62]. During mitophagy, the extraneous or defective mitochondria are trafficked to the lysosome, where they are degraded by lysosomal enzymes [
63]. Various animal and human studies have established the role of impaired mitophagy in AD [
64]. Dysfunctional mitochondria lead to the accumulation of excess ROS and the depletion of ATP [
65,
66]. Impaired mitochondria at distal sites have to be transported to the soma for lysosomal degradation, and this retrograde transport may be impaired in AD [
67,
68].
4.2. Aβ and Tau in Mitophagy and Mitochondrial Movement
The most well-studied causes of mitochondrial dysfunction in AD relate to the toxicity of Aβ and tau. The accumulation of Aβ causes oxidative stress and the production of ROS by mitochondria. The ROS generated then inflicts damage on mitochondria [
70,
71]. Mitochondrial ROS production promotes tau aggregation [
72]. Aβ and tau also interfere with the trafficking of mitochondria to and from the synapse while also fostering mitochondrial fission, leading to synaptic dysfunction [
73,
74].
Aβ is not produced locally in the mitochondria, so mitochondrial Aβ uptake poses an interesting area of study. Petersen et al. found that in rat mitochondria, Aβ is transported via the translocase of the outer membrane machinery [
75]. Immunoelectron microscopy after import showed localization of Aβ to mitochondrial cristae. This was similarly found in human cortical brain biopsies, suggesting that this import machinery can be a unique mechanism for Aβ entry into mitochondria.
Dou and Tan transfected SHSY-5Y human neuroblastoma cells with plasmids harboring mitochondrial outer membrane protein translocase (TOMM)22 and TOMM40 to directly augment mitochondrial Aβ content. They found that increased Aβ content in the mitochondria enhanced mitophagy, and this could be reversed by transfection with a plasmid harboring presequence protease, responsible for Aβ degradation [
76].
The tau protein, a microtubule-associated protein important for synaptic plasticity, acts as a promoter of microtubule assembly, a microtubule stabilizer, and an autophagy regulator [
78,
79]. The tau protein is primarily expressed in neurons, where it is involved in axonal transport [
80,
81]. Accumulation of misfolded insoluble phosphorylated tau protein in the neuron leads to aggregation into neurofibrillary tangles that are neurotoxic [
82,
83,
84]. Hyperphosphorylation of tau protein reduces its binding affinity to microtubules, causing microtubules to dissemble and interfering with axonal transport of mitochondria and synaptic vesicles [
85,
86]. Insufficient mitochondrial presence along the axon starves the synapse of ATP and energy and impedes the autophagic clearance of mitochondria in neurons [
87,
88].
Hu et al. examined the association between intracellular tau accumulation, a hallmark of sporadic AD, and mitophagy using the mitochondrial marker proteins cytochrome c oxidase (COX) IV and TOMM20 [
89]. Comparing brain homogenates from AD subjects and age-matched controls, Western blotting showed higher levels of COX IV, TOMM20, total tau and phosphorylated tau in the AD patients. Interestingly, only AD subjects with high tau levels had elevations in COX IV and TOMM20, while AD subjects with normal total tau levels had COX IV and TOMM20 expression comparable to non-AD controls. Since high levels of COX IV and TOMM20 may be considered indicators of mitophagy deficits, these results suggest an association between intracellular tau accumulation and mitophagy deficits [
90].
4.3. Mitochondrial Fission and Fusion
Mitochondrial fission and fusion are both controlled by large guanosine triphosphatases (GTPases) in the dynamin family. The balance between fusion and fission is critical for meeting energy demand, as excess fission leads to mitochondrial fragmentation while excess fusion leads to elongated mitochondria with high levels of ROS [
93]. The specific proteins that regulate fission include dynamin-1-related protein (DRP)1, fission (Fis)1, and mitochondrial fission factor (Mff), while mitochondrial fusion is regulated by proteins such as mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy protein (OPA1) [
94].
While mitochondrial fission is required for both mitophagy and mitochondrial transport, excess fission leads to fragmentation, and patients with AD are determined to have higher expression of mitochondrial fission genes such as DRP1 [
95,
96,
97,
98]. Enhanced fission leads to structural damage to mitochondria in neurons in the AD brain [
99,
100]. Upregulation of fusion proteins or interference with fission proteins may rescue neurons from the consequences of overzealous fission [
101,
102,
103].
4.4. Effects of Amyloid and Tau on Fission and Fusion
Drp1 may contribute to the pathogenesis of AD by interacting with Aβ and phosphorylated tau, leading to excessive mitochondrial fragmentation with negative consequences such as synaptic dysfunction and neuronal damage [
105].
Glycogen synthase kinase 3 β (GSK3β), primarily present in the brain and activated by Aβ, is responsible for phosphorylation of tau and is considered a crucial enzyme in the pathobiology of AD [
106,
107]. In murine models, overexpression of GSK3β increases tau phosphorylation and promotes disassembly of microtubules [
108]. GSK3β also phosphorylates Drp1 at multiple serines, affecting mitochondrial fission and fragmentation [
109,
110].
5. Mitochondrial DNA Methylation
Mitochondrial DNA methylation, a mechanism of epigenetic control, is a suspected contributing factor in AD pathogenesis [
117,
118]. Xu and colleagues compared mitochondrial DNA methylation in the hippocampi of transgenic APP/PS1 AD mice to age-matched wild-type C57BL/6J mice and found hypomethylation of the D-loop region (critical for mitochondrial DNA replication and transcription) and hypermethylation of the 12 S rRNA gene in the hippocampi of the AD mouse model [
119]. The AD mice also showed a decrease in mitochondrial DNA copy number and lower gene expression compared to the C57BL/6J mice, indicative of mitochondrial dysfunction and abnormal biogenesis [
120,
121].
6. Glucose Metabolism Reduced in AD
The human brain is one of the most metabolically active organs in the body and predominantly utilizes glucose as its main energy source. The brain is relatively inflexible in using alternative substrates aside from glucose for energy production [
127,
128]. AD is characterized by reduced glucose metabolism in the brain [
129,
130]. This hypometabolism is detectable in PET scans and serves as an early imaging modality for AD detection and prediction of progression from MCI to AD [
131,
132,
133,
134]. A longitudinal study of cognitively normal older persons and persons with mild AD showed a progressive reduction in glucose utilization prior to dementia onset in those who began the study without cognitive symptoms [
135]. In the early stages of AD, glucose hypometabolism is apparent in the hippocampal and posterior cingulate of the human brain, areas that also show abnormal patterns of functional connectivity early in AD [
136,
137,
138]. As the disease advances, glucose consumption is reduced at the temporal–parietal cortex and the frontal and occipital cortices [
139]. Hypometabolism is generally bilateral in AD but may be left lateralized in early MCI [
140].
Hypometabolism may be due partly to reduced glucose transport at the blood–brain barrier and across astrocytic and neuronal cell membranes. The transport of glucose from the bloodstream to the parenchymal cells is facilitated by integral membrane proteins called glucose transporters (GLUTs). These sodium-independent facilitative transporters play an important role in glucose metabolism [
141]. The majority of glucose uptake in the brain occurs via GLUT1 and GLUT3. While GLUT1 moves glucose across the blood–brain barrier into astrocytes, Glut3 handles the majority of glucose uptake by neurons [
142,
143]. Decreased levels of GLUT1 and GLUT3 are particularly seen in the cerebral cortex and hippocampus of AD patients, with significant loss of GLUT3 [
144]. One proposed mechanism for decreased levels of GLUT1 and GLUT3 in the AD brain is the downregulation of hypoxia-inducible factor-1 (HIF-1) [
145]. HIF-1 suppression subsequently causes abnormal tau phosphorylation and/or neurofibrillary degeneration by downregulating the hexosamine biosynthesis pathway. [
146]. Another putative mechanism contributing to decreased GLUT3 expression involves the transcription factor cAMP response element (CRE)-binding protein (CREB), known to be important in supporting cognition, memory formation, and neuronal survival [
147]. The human GLUT3 promoter has three potential (CRE)-like elements where CREB can bind and induce GLUT3 expression.
7. Apolipoprotein (Apo)E Gene Impact on Mitochondria and Bioenergetics
ApoE is a 299 amino acid lipid transport protein and the most abundant brain apolipoprotein. ApoE functions to maintain brain lipid balance and facilitates the exchange of lipids between neurons and glial cells. Its expression in the brain is upregulated in activated microglia and in stressed neurons as an adaptation to inflammatory and cellular stress conditions [
160,
161].
There are three predominant ApoE isoforms in humans: ApoE2, ApoE3, and ApoE4, which are the products of the ε2, ε3, and ε4 alleles, respectively. These confer varying degrees of AD risk, with the ApoE ε4 allele being the strongest genetic risk factor for sporadic AD, while the ApoE ε2 allele is associated with the lowest AD risk [
162,
163,
164]. ApoE4 is expressed in more than half of AD patients, and its prevalence makes it an important therapeutic target [
165,
166].
8. Mitochondria in the Treatment of AD
The importance of mitochondrial dysfunction in the pathogenesis of AD has prompted the exploration of new treatment strategies designed to improve mitochondrial function [
178,
179,
180]. A major focus has been on correcting oxidative stress imbalances using mitochondrial-targeted antioxidant therapies, and many studies have been conducted in rodent models showing efficacy [
181,
182,
183].
Mitoquinone mesylate (MitoQ, 10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadienlyl) decyl triphenylphosphonium methanesulfonate)) is a compound composed of a derivative of ubiquinone targeted to mitochondria by covalent attachment to a lipophilic triphenylphosphonium, which facilitates crossing of the molecule through the layers of the mitochondrial membranes. The ubiquinone can then be converted to the antioxidant ubiquinol by complex II of the ETC. MitoQ behaves as a scavenger for ROS and has been previously tested in AD nematode and mouse model systems [
184,
185,
186].
Other antioxidant compounds such as mito-apocynin, made from apocynin, a plant-derived inhibitor of NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, and astaxanthin, a red pigment with potent antioxidant properties, have also shown potential for improving mitochondrial dysfunction in preclinical models and could be used in humans in the future [
189,
190,
191,
192,
193].
Mitochondrial fragmentation is detrimental to cellular bioenergetics. As discussed earlier, the mitochondrial fission protein Drp1 is abnormally expressed in AD, leading to excess mitochondrial fragmentation [
194]. Drp1 also interacts with Aβ and hyperphosphorylated tau and promotes changes in mitochondrial morphology and bioenergetics, negatively impacting ATP production. Interactions between Drp1 and Aβ induce synaptic loss [
195,
196]. The Drp1 inhibitor, mitochondria division inhibitor 1 (Mdivi1), a quinazolinone derivative, has been found to effectively target synaptic depression that occurs due to Aβ in AD [
197,
198]. Mdivi1 has been found to specifically target mitochondrial dysfunction by attenuating ROS production and enhancing ATP production [
199]. These findings suggest that Mdivi1 is a potential therapeutic option for treating mitochondrial dysfunction and synaptic depression associated with Aβ-induced pathology in hippocampal cells in AD [
200,
201].
Diethyl (3,4-dihydroxyphenethylamino) (quinoline-4-yl)methyl phosphonate (DDQ) is a pharmacologically developed compound that can cross the blood–brain barrier and has shown positive effects on mitochondrial dysfunction and synaptic dysregulation at both mRNA and protein levels [
203,
204]. DDQ reduces the fission proteins Drp1 and Fis1 while increasing the fusion proteins Mfn1 and Mfn2. It reduces the interactions of DRP1 with Aβ, inhibiting Aβ-DRP1 complex formations of [
205]. Aβ-Drp1 complexes are known to promote mitochondrial fragmentation, mitochondrial DNA mutations, and a reduction in mitochondrial oxidative phosphorylation, all of which are observed in AD brains. Drp1 inhibition has emerged as a therapeutic target because it has been shown to improve learning and memory and protect mitochondria from fragmentation in mouse models of AD [
206,
207].
While studies are ongoing to find breakthrough treatments for AD, maintaining a healthy lifestyle and optimizing cognitive reserve can improve quality of life and perhaps delay the symptomatic phase of AD
[2].
10. Conclusions
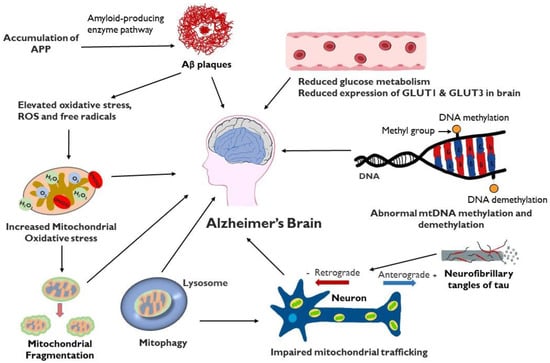
Mitochondrial dysfunction plays a critical role in multiple aspects of the development of AD (Figure 2). Failure of mitochondria leads to insufficient energy supply and oxidative stress, which further erodes mitochondrial integrity and damages the neuron, particularly the axon. Restoring and maintaining mitochondrial health is increasingly the focus of investigation as a therapeutic strategy in AD, particularly in light of the inability of anti-amyloid and anti-tau treatments to halt AD progression.
Figure 2. Schematic representation of factors involved in mitochondrial dysfunction and AD: Mitochondrial dysfunction in the AD brain results in elevated oxidative stress, increased mitochondrial fragmentation, mitophagy, impaired mitochondrial trafficking, mitochondrial DNA damage, defective mitochondrial biogenesis and dynamics and reduced glucose metabolism. Accumulation of Aβ can contribute to mitochondrial oxidative stress while tau protein, present in extracellular tangles, can interfere with axonal movement of mitochondria.
This entry is adapted from the peer-reviewed paper 10.3390/life14020196