 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudia Tanja Mierke | -- | 7961 | 2024-02-08 15:29:09 | | | |
| 2 | Mona Zou | -2 word(s) | 7959 | 2024-02-09 10:20:31 | | |
Video Upload Options
Tumor diseases become a huge problem when they embark on a path that advances to malignancy, such as the process of metastasis. Cancer metastasis has been thoroughly investigated from a biological perspective in the past, whereas it has still been less explored from a physical perspective. Until now, the intraluminal pathway of cancer metastasis has received the most attention, while the interaction of cancer cells with macrophages has received little attention. Apart from the biochemical characteristics, tumor treatments also rely on the tumor microenvironment, which is recognized to be immunosuppressive and, as has recently been found, mechanically stimulates cancer cells and thus alters their functions.
1. Cancer Development
1.1. Driver Mutations Advance Cancerogenesis
1.2. Establishment of Basic Primary Cancer Models
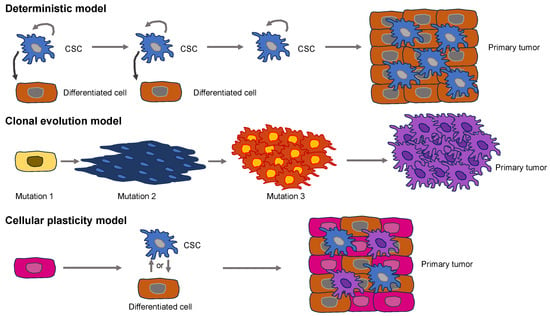
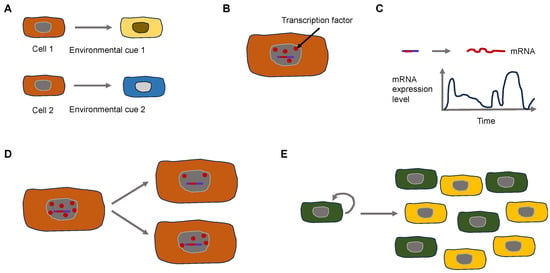
2. Universal Properties of Cancer and Their Effect on Cancer Cell Function
3. Malignant Cancer Progression (Cancer Metastasis)
3.1. Increased Vascular Permeability around Cancers
3.2. Bodily Fluids
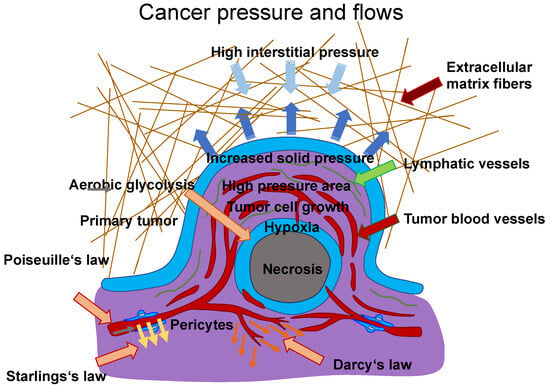
3.3. Tumor Interstitial Fluid Facilitates the Cancer Cell’s Migration and Invasion
(Figure 3, gray arrow) to the radium of the vessel R, the dynamic blood fluid viscosity µ, and the intravascular pressure gradient 𝑝𝑣. Starling’s law (Equation (2)) links the rate of extravasation 𝐽𝑣 (Figure 3, yellow arrows) to the hydraulic conductivity of the vascular wall 𝐿𝑝, the vascular surface area S, the reflection coefficient σ, the vascular oncotic pressure 𝜋𝑣, and the interstitial oncotic pressure 𝜋𝑖. Darcy’s law (Equation (3)) connects the interstitial flow speed 𝑢𝑡 (Figure 3, orange arrows) to the interstitial tissue hydraulic conductivity 𝑘 and the interstitial pressure gradient 𝑝𝑡. These three linkages can be found within the overall published literature on the physical modeling of tumor-based vascular flux and angiogenesis.
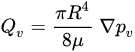
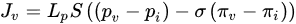
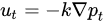
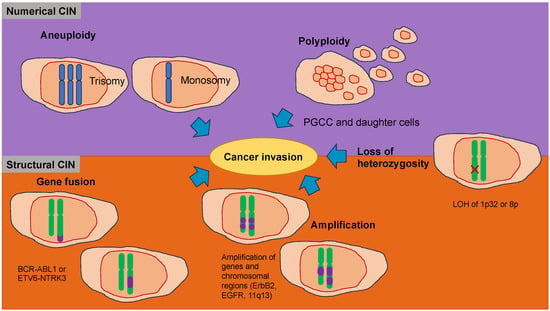
4. Chromosomal Instability, Exosomes, and Cell-Free DNA Foster Cancer Metastasis
4.1. Chromosomal Instability
4.2. Exosomes
4.3. Cell-Free DNA (cfDNA)
References
- Lynch, M. Rate, Molecular Spectrum, and Consequences of Human Mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968.
- Armitage, P.; Doll, R. The Age Distribution of Cancer and a Multi-Stage Theory of Carcinogenesis. Br. J. Cancer 1954, 8, 1–12.
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of Somatic Mutation in Human Cancer Genomes. Nature 2007, 446, 153–158.
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only Three Driver Gene Mutations Are Required for the Development of Lung and Colorectal Cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123.
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886.
- St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253.
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A.; Papadopoulos, N.; et al. Integrated Genomic Analyses Identify ARID1A and ARID1B Alterations in the Childhood Cancer Neuroblastoma. Nat. Genet. 2013, 45, 12–17.
- Espiritu, D.; Gribkova, A.K.; Gupta, S.; Shaytan, A.K.; Panchenko, A.R. Molecular Mechanisms of Oncogenesis through the Lens of Nucleosomes and Histones. J. Phys. Chem. B 2021, 125, 3963–3976.
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic Heterogeneity in Cancer. Biomark. Res. 2019, 7, 23.
- Sadri Nahand, J.; Rabiei, N.; Fathazam, R.; Taghizadieh, M.; Ebrahimi, M.S.; Mahjoubin-Tehran, M.; Bannazadeh Baghi, H.; Khatami, A.; Abbasi-Kolli, M.; Mirzaei, H.R.; et al. Oncogenic Viruses and Chemoresistance: What Do We Know? Pharmacol. Res. 2021, 170, 105730.
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151.
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and Cancer Stem Cells: Molecular Mechanisms and Therapeutic Applications. Cell Death Differ. 2019, 26, 690–702.
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of Cancer Stem Cells (CSCs) and Mechanisms of Their Regulation: Implications for Cancer Therapy. Curr. Protoc. Pharmacol. 2013, 61, 14–25.
- Rossi, F.; Noren, H.; Jove, R.; Beljanski, V.; Grinnemo, K.-H. Differences and Similarities between Cancer and Somatic Stem Cells: Therapeutic Implications. Stem Cell Res. Ther. 2020, 11, 489.
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313.
- Williams, M.J.; Sottoriva, A.; Graham, T.A. Measuring Clonal Evolution in Cancer with Genomics. Annu. Rev. Genom. Hum. Genet. 2019, 20, 309–329.
- Shlyakhtina, Y.; Moran, K.L.; Portal, M.M. Genetic and Non-Genetic Mechanisms Underlying Cancer Evolution. Cancers 2021, 13, 1380.
- Shen, S.; Clairambault, J. Cell Plasticity in Cancer Cell Populations. F1000Research 2020, 9, 635.
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging Role of Tumor Cell Plasticity in Modifying Therapeutic Response. Signal Transduct. Target. Ther. 2020, 5, 228.
- Deshmukh, S.; Saini, S. Phenotypic Heterogeneity in Tumor Progression, and Its Possible Role in the Onset of Cancer. Front. Genet. 2020, 11, 604528.
- Huh, D.; Paulsson, J. Non-Genetic Heterogeneity from Stochastic Partitioning at Cell Division. Nat. Genet. 2011, 43, 95–100.
- Huh, D.; Paulsson, J. Random Partitioning of Molecules at Cell Division. Proc. Natl. Acad. Sci. USA 2011, 108, 15004–15009.
- Swain, P.S.; Elowitz, M.B.; Siggia, E.D. Intrinsic and Extrinsic Contributions to Stochasticity in Gene Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 12795–12800.
- Mierke, C.T.; Frey, B.; Fellner, M.; Herrmann, M.; Fabry, B. Integrin A5β1 Facilitates Cancer Cell Invasion through Enhanced Contractile Forces. J. Cell Sci. 2011, 124, 369–383.
- Sotodosos-Alonso, L.; Pulgarín-Alfaro, M.; Del Pozo, M.A. Caveolae Mechanotransduction at the Interface between Cytoskeleton and Extracellular Matrix. Cells 2023, 12, 942.
- Grande-García, A.; Echarri, A.; De Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; Del Pozo, M.A. Caveolin-1 Regulates Cell Polarization and Directional Migration through Src Kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694.
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical Remodeling of the Microenvironment by Stromal Caveolin-1 Favors Tumor Invasion and Metastasis. Cell 2011, 146, 148–163.
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635.e6.
- Nikolić, M.; Scarcelli, G.; Tanner, K. Multimodal Microscale Mechanical Mapping of Cancer Cells in Complex Microenvironments. Biophys. J. 2022, 121, 3586–3599.
- Lin, H.-H.; Lin, H.-K.; Lin, I.-H.; Chiou, Y.-W.; Chen, H.-W.; Liu, C.-Y.; Harn, H.I.-C.; Chiu, W.-T.; Wang, Y.-K.; Shen, M.-R.; et al. Mechanical Phenotype of Cancer Cells: Cell Softening and Loss of Stiffness Sensing. Oncotarget 2015, 6, 20946–20958.
- Wang, H.-B.; Dembo, M.; Wang, Y.-L. Substrate Flexibility Regulates Growth and Apoptosis of Normal but Not Transformed Cells. Am. J. Physiol.-Cell Physiol. 2000, 279, C1345–C1350.
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689.
- Walser, P.J.; Ariotti, N.; Howes, M.; Ferguson, C.; Webb, R.; Schwudke, D.; Leneva, N.; Cho, K.-J.; Cooper, L.; Rae, J.; et al. Constitutive Formation of Caveolae in a Bacterium. Cell 2012, 150, 752–763.
- Parton, R.G. Caveolae: Structure, Function, and Relationship to Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 111–136.
- Cheng, J.P.X.; Nichols, B.J. Caveolae: One Function or Many? Trends Cell Biol. 2016, 26, 177–189.
- Arpaia, E.; Blaser, H.; Quintela-Fandino, M.; Duncan, G.; Leong, H.S.; Ablack, A.; Nambiar, S.C.; Lind, E.F.; Silvester, J.; Fleming, C.K.; et al. The Interaction between Caveolin-1 and Rho-GTPases Promotes Metastasis by Controlling the Expression of Alpha5-Integrin and the Activation of Src, Ras and Erk. Oncogene 2012, 31, 884–896.
- Nunez-Wehinger, S.; Ortiz, R.J.; Diaz, N.; Diaz, J.; Lobos-Gonzalez, L.; Quest, A.F.G. Caveolin-1 in Cell Migration and Metastasis. Curr. Mol. Med. 2014, 14, 255–274.
- Torres, V.A.; Tapia, J.C.; Rodriguez, D.A.; Lladser, A.; Arredondo, C.; Leyton, L.; Quest, A.F.G. E-Cadherin Is Required for Caveolin-1-Mediated Down-Regulation of the Inhibitor of Apoptosis Protein Survivin via Reduced β-Catenin-Tcf/Lef-Dependent Transcription. Mol. Cell. Biol. 2007, 27, 7703–7717.
- Rodriguez, D.A.; Tapia, J.C.; Fernandez, J.G.; Torres, V.A.; Muñoz, N.; Galleguillos, D.; Leyton, L.; Quest, A.F.G. Caveolin-1–Mediated Suppression of Cyclooxygenase-2 via a β-Catenin-Tcf/Lef–Dependent Transcriptional Mechanism Reduced Prostaglandin E 2 Production and Survivin Expression. Mol. Biol. Cell 2009, 20, 2297–2310.
- Quest, A.F.G.; Gutierrez-Pajares, J.L.; Torres, V.A. Caveolin-1: An Ambiguous Partner in Cell Signalling and Cancer. J. Cell. Mol. Med. 2008, 12, 1130–1150.
- Gupta, V.K.; Sharma, N.S.; Kesh, K.; Dauer, P.; Nomura, A.; Giri, B.; Dudeja, V.; Banerjee, S.; Bhattacharya, S.; Saluja, A.; et al. Metastasis and Chemoresistance in CD133 Expressing Pancreatic Cancer Cells Are Dependent on Their Lipid Raft Integrity. Cancer Lett. 2018, 439, 101–112.
- Teo, J.L.; Gomez, G.A.; Weeratunga, S.; Davies, E.M.; Noordstra, I.; Budnar, S.; Katsuno-Kambe, H.; McGrath, M.J.; Verma, S.; Tomatis, V.; et al. Caveolae Control Contractile Tension for Epithelia to Eliminate Tumor Cells. Dev. Cell 2020, 54, 75–91.e7.
- Charras, G.; Yap, A.S. Tensile Forces and Mechanotransduction at Cell–Cell Junctions. Curr. Biol. 2018, 28, R445–R457.
- Reymann, A.-C.; Staniscia, F.; Erzberger, A.; Salbreux, G.; Grill, S.W. Cortical Flow Aligns Actin Filaments to Form a Furrow. eLife 2016, 5, e17807.
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627.
- Schönichen, A.; Geyer, M. Fifteen Formins for an Actin Filament: A Molecular View on the Regulation of Human Formins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 152–163.
- Grikscheit, K.; Grosse, R. Formins at the Junction. Trends Biochem. Sci. 2016, 41, 148–159.
- Nassoy, P.; Lamaze, C. Stressing Caveolae New Role in Cell Mechanics. Trends Cell Biol. 2012, 22, 381–389.
- Lamaze, C.; Torrino, S. Caveolae and Cancer: A New Mechanical Perspective. Biomed. J. 2015, 38, 367.
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell 2011, 144, 402–413.
- Cheng, J.P.X.; Mendoza-Topaz, C.; Howard, G.; Chadwick, J.; Shvets, E.; Cowburn, A.S.; Dunmore, B.J.; Crosby, A.; Morrell, N.W.; Nichols, B.J. Caveolae Protect Endothelial Cells from Membrane Rupture during Increased Cardiac Output. J. Cell Biol. 2015, 211, 53–61.
- Lo, H.P.; Nixon, S.J.; Hall, T.E.; Cowling, B.S.; Ferguson, C.; Morgan, G.P.; Schieber, N.L.; Fernandez-Rojo, M.A.; Bastiani, M.; Floetenmeyer, M.; et al. The Caveolin–Cavin System Plays a Conserved and Critical Role in Mechanoprotection of Skeletal Muscle. J. Cell Biol. 2015, 210, 833–849.
- Dewulf, M.; Köster, D.V.; Sinha, B.; Viaris De Lesegno, C.; Chambon, V.; Bigot, A.; Bensalah, M.; Negroni, E.; Tardif, N.; Podkalicka, J.; et al. Dystrophy-Associated Caveolin-3 Mutations Reveal That Caveolae Couple IL6/STAT3 Signaling with Mechanosensing in Human Muscle Cells. Nat. Commun. 2019, 10, 1974.
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in Oncogenic Transformation, Cancer, and Metastasis. Am. J. Physiol.-Cell Physiol. 2005, 288, C494–C506.
- Chatterjee, M.; Ben-Josef, E.; Thomas, D.G.; Morgan, M.A.; Zalupski, M.M.; Khan, G.; Andrew Robinson, C.; Griffith, K.A.; Chen, C.-S.; Ludwig, T.; et al. Caveolin-1 Is Associated with Tumor Progression and Confers a Multi-Modality Resistance Phenotype in Pancreatic Cancer. Sci. Rep. 2015, 5, 10867.
- Simón, L.; Campos, A.; Leyton, L.; Quest, A.F.G. Caveolin-1 Function at the Plasma Membrane and in Intracellular Compartments in Cancer. Cancer Metastasis Rev. 2020, 39, 435–453.
- Porter, R.J.; Murray, G.I.; McLean, M.H. Current Concepts in Tumour-Derived Organoids. Br. J. Cancer 2020, 123, 1209–1218.
- Tatullo, M.; Marrelli, B.; Benincasa, C.; Aiello, E.; Makeeva, I.; Zavan, B.; Ballini, A.; De Vito, D.; Spagnuolo, G. Organoids in Translational Oncology. J. Clin. Med. 2020, 9, 2774.
- Mierke, C.T. Physical and Biological Advances in Endothelial Cell-Based Engineered Co-Culture Model Systems. Semin. Cell Dev. Biol. 2023, 147, 58–69.
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog Signaling Induces PD-L1 Expression and Tumor Cell Proliferation in Gastric Cancer. Oncotarget 2018, 9, 37439–37457.
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.; Chang, J.; Dlugosz, A.; Zavros, Y. Mouse-Derived Gastric Organoid and Immune Cell Co-Culture for the Study of the Tumor Microenvironment. In Epithelial Cell Culture; Baratta, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1817, pp. 157–168. ISBN 978-1-4939-8599-9.
- Suarez, G.; Romero-Gallo, J.; Piazuelo, M.B.; Sierra, J.C.; Delgado, A.G.; Washington, M.K.; Shah, S.C.; Wilson, K.T.; Peek, R.M. Nod1 Imprints Inflammatory and Carcinogenic Responses toward the Gastric Pathogen Helicobacter Pylori. Cancer Res. 2019, 79, 1600–1611.
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes Activate Mitochondrial Fatty Acid Oxidation and Autophagy to Promote Tumor Growth in Colon Cancer. Cell Death Dis. 2017, 8, e2593.
- Curran, S.; Murray, G.I. Matrix Metalloproteinases in Tumour Invasion and Metastasis. J. Pathol. 1999, 189, 300–308.
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement Membrane Destruction by Pancreatic Stellate Cells Leads to Local Invasion in Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2018, 425, 65–77.
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of Primary Human Pancreatic Cancer Organoids, Matched Stromal and Immune Cells and 3D Tumor Microenvironment Models. BMC Cancer 2018, 18, 335.
- Mollica, P.A.; Booth-Creech, E.N.; Reid, J.A.; Zamponi, M.; Sullivan, S.M.; Palmer, X.-L.; Sachs, P.C.; Bruno, R.D. 3D Bioprinted Mammary Organoids and Tumoroids in Human Mammary Derived ECM Hydrogels. Acta Biomater. 2019, 95, 201–213.
- Usui, T.; Sakurai, M.; Umata, K.; Yamawaki, H.; Ohama, T.; Sato, K. Preparation of Human Primary Colon Tissue-Derived Organoid Using Air Liquid Interface Culture. Curr. Protoc. Toxicol. 2018, 75, 22–26.
- Alibert, C.; Goud, B.; Manneville, J.-B. Are Cancer Cells Really Softer than Normal Cells?: Mechanics of Cancer Cells. Biol. Cell 2017, 109, 167–189.
- Li, M.; Dang, D.; Liu, L.; Xi, N.; Wang, Y. Atomic Force Microscopy in Characterizing Cell Mechanics for Biomedical Applications: A Review. IEEE Trans. Nanobiosci. 2017, 16, 523–540.
- Zemła, J.; Danilkiewicz, J.; Orzechowska, B.; Pabijan, J.; Seweryn, S.; Lekka, M. Atomic Force Microscopy as a Tool for Assessing the Cellular Elasticity and Adhesiveness to Identify Cancer Cells and Tissues. Semin. Cell Dev. Biol. 2018, 73, 115–124.
- Efremov, Y.M.; Shimolina, L.; Gulin, A.; Ignatova, N.; Gubina, M.; Kuimova, M.K.; Timashev, P.S.; Shirmanova, M.V. Correlation of Plasma Membrane Microviscosity and Cell Stiffness Revealed via Fluorescence-Lifetime Imaging and Atomic Force Microscopy. Cells 2023, 12, 2583.
- Swaminathan, V.; Mythreye, K.; O’Brien, E.T.; Berchuck, A.; Blobe, G.C.; Superfine, R. Mechanical Stiffness Grades Metastatic Potential in Patient Tumor Cells and in Cancer Cell Lines. Cancer Res. 2011, 71, 5075–5080.
- Xu, W.; Mezencev, R.; Kim, B.; Wang, L.; McDonald, J.; Sulchek, T. Cell Stiffness Is a Biomarker of the Metastatic Potential of Ovarian Cancer Cells. PLoS ONE 2012, 7, e46609.
- Watanabe, T.; Kuramochi, H.; Takahashi, A.; Imai, K.; Katsuta, N.; Nakayama, T.; Fujiki, H.; Suganuma, M. Higher Cell Stiffness Indicating Lower Metastatic Potential in B16 Melanoma Cell Variants and in (−)-Epigallocatechin Gallate-Treated Cells. J. Cancer Res. Clin. Oncol. 2012, 138, 859–866.
- Zhang, G.; Long, M.; Wu, Z.-Z.; Yu, W.-Q. Mechanical Properties of Hepatocellular Carcinoma Cells. World J. Gastroenterol. 2002, 8, 243.
- Faria, E.C.; Ma, N.; Gazi, E.; Gardner, P.; Brown, M.; Clarke, N.W.; Snook, R.D. Measurement of Elastic Properties of Prostate Cancer Cells Using AFM. Analyst 2008, 133, 1498.
- Staunton, J.R.; Doss, B.L.; Lindsay, S.; Ros, R. Correlating Confocal Microscopy and Atomic Force Indentation Reveals Metastatic Cancer Cells Stiffen during Invasion into Collagen I Matrices. Sci. Rep. 2016, 6, 19686.
- Molter, C.W.; Muszynski, E.F.; Tao, Y.; Trivedi, T.; Clouvel, A.; Ehrlicher, A.J. Prostate Cancer Cells of Increasing Metastatic Potential Exhibit Diverse Contractile Forces, Cell Stiffness, and Motility in a Microenvironment Stiffness-Dependent Manner. Front. Cell Dev. Biol. 2022, 10, 932510.
- Fischer, T.; Wilharm, N.; Hayn, A.; Mierke, C.T. Matrix and Cellular Mechanical Properties Are the Driving Factors for Facilitating Human Cancer Cell Motility into 3D Engineered Matrices. Converg. Sci. Phys. Oncol. 2017, 3, 044003.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Kihara, T.; Haghparast, S.M.A.; Shimizu, Y.; Yuba, S.; Miyake, J. Physical Properties of Mesenchymal Stem Cells Are Coordinated by the Perinuclear Actin Cap. Biochem. Biophys. Res. Commun. 2011, 409, 1–6.
- Efremov, Y.M.; Velay-Lizancos, M.; Weaver, C.J.; Athamneh, A.I.; Zavattieri, P.D.; Suter, D.M.; Raman, A. Anisotropy vs Isotropy in Living Cell Indentation with AFM. Sci. Rep. 2019, 9, 5757.
- Massagué, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306.
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov. 2017, 7, 1224–1237.
- Mohammadi, H.; Sahai, E. Mechanisms and Impact of Altered Tumour Mechanics. Nat. Cell Biol. 2018, 20, 766–774.
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534.
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The Physics of Cancer: The Role of Physical Interactions and Mechanical Forces in Metastasis. Nat. Rev. Cancer 2011, 11, 512–522.
- Swartz, M.A.; Lund, A.W. Lymphatic and Interstitial Flow in the Tumour Microenvironment: Linking Mechanobiology with Immunity. Nat. Rev. Cancer 2012, 12, 210–219.
- Koumoutsakos, P.; Pivkin, I.; Milde, F. The Fluid Mechanics of Cancer and Its Therapy. Annu. Rev. Fluid Mech. 2013, 45, 325–355.
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and Growth of Cancer Cells in Metastatic Sites. Nat. Rev. Cancer 2002, 2, 563–572.
- Joyce, J.A.; Pollard, J.W. Microenvironmental Regulation of Metastasis. Nat. Rev. Cancer 2009, 9, 239–252.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; Van De Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of Lymphatic and Distant Metastases in Human Colorectal Cancer. Science 2017, 357, 55–60.
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph Node Blood Vessels Provide Exit Routes for Metastatic Tumor Cell Dissemination in Mice. Science 2018, 359, 1408–1411.
- Pereira, E.R.; Kedrin, D.; Seano, G.; Gautier, O.; Meijer, E.F.J.; Jones, D.; Chin, S.-M.; Kitahara, S.; Bouta, E.M.; Chang, J.; et al. Lymph Node Metastases Can Invade Local Blood Vessels, Exit the Node, and Colonize Distant Organs in Mice. Science 2018, 359, 1403–1407.
- Weiss, L.; Bronk, J.; Pickren, J.W.; Lane, W.W. Metastatic Patterns and Target Organ Arterial Blood Flow. Invasion Metastasis 1981, 1, 126–135.
- Headley, M.B.; Bins, A.; Nip, A.; Roberts, E.W.; Looney, M.R.; Gerard, A.; Krummel, M.F. Visualization of Immediate Immune Responses to Pioneer Metastatic Cells in the Lung. Nature 2016, 531, 513–517.
- Follain, G.; Osmani, N.; Azevedo, A.S.; Allio, G.; Mercier, L.; Karreman, M.A.; Solecki, G.; Garcia Leòn, M.J.; Lefebvre, O.; Fekonja, N.; et al. Hemodynamic Forces Tune the Arrest, Adhesion, and Extravasation of Circulating Tumor Cells. Dev. Cell 2018, 45, 33–52.e12.
- Huang, Q.; Li, S.; Hu, X.; Sun, M.; Wu, Q.; Dai, H.; Tan, Y.; Sun, F.; Wang, C.; Rong, X.; et al. Shear Stress Activates ATOH8 via Autocrine VEGF Promoting Glycolysis Dependent-Survival of Colorectal Cancer Cells in the Circulation. J. Exp. Clin. Cancer Res. 2020, 39, 25.
- Egginton, S. Physiological Factors Influencing Capillary Growth. Acta Physiol. 2011, 202, 225–239.
- Jain, R.K. Determinants of Tumor Blood Flow: A Review. Cancer Res. 1988, 48, 2641–2658.
- Jain, R.K. Vascular and Interstitial Barriers to Delivery of Therapeutic Agents in Tumors. Cancer Metastasis Rev. 1990, 9, 253–266.
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of Vascular Normalization by Antiangiogenic Therapy on Interstitial Hypertension, Peritumor Edema, and Lymphatic Metastasis: Insights from a Mathematical Model. Cancer Res. 2007, 67, 2729–2735.
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121.
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat. Commun. 2013, 4, 2516.
- Maeda, H. The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: The Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzym. Regul. 2001, 41, 189–207.
- Freund, J.B. Numerical Simulation of Flowing Blood Cells. Annu. Rev. Fluid Mech. 2014, 46, 67–95.
- Bessonov, N.; Sequeira, A.; Simakov, S.; Vassilevskii, Y.; Volpert, V. Methods of Blood Flow Modelling. Math. Model. Nat. Phenom. 2016, 11, 1–25.
- Dixon, J.B.; Greiner, S.T.; Gashev, A.A.; Cote, G.L.; Moore, J.E.; Zawieja, D.C. Lymph Flow, Shear Stress, and Lymphocyte Velocity in Rat Mesenteric Prenodal Lymphatics. Microcirculation 2006, 13, 597–610.
- Peng, S.-L.; Shih, C.-T.; Huang, C.-W.; Chiu, S.-C.; Shen, W.-C. Optimized Analysis of Blood Flow and Wall Shear Stress in the Common Carotid Artery of Rat Model by Phase-Contrast MRI. Sci. Rep. 2017, 7, 5253.
- Reneman, R.S.; Hoeks, A.P.G. Wall Shear Stress as Measured in Vivo: Consequences for the Design of the Arterial System. Med. Biol. Eng. Comput. 2008, 46, 499–507.
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319.
- Levick, J.R.; Michel, C.C. Microvascular Fluid Exchange and the Revised Starling Principle. Cardiovasc. Res. 2010, 87, 198–210.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and Lymphatic Vessel Remodelling in Cancer. Nat. Rev. Cancer 2014, 14, 159–172.
- Broggi, M.A.S.; Maillat, L.; Clement, C.C.; Bordry, N.; Corthésy, P.; Auger, A.; Matter, M.; Hamelin, R.; Potin, L.; Demurtas, D.; et al. Tumor-Associated Factors Are Enriched in Lymphatic Exudate Compared to Plasma in Metastatic Melanoma Patients. J. Exp. Med. 2019, 216, 1091–1107.
- Baish, J.W.; Netti, P.A.; Jain, R.K. Transmural Coupling of Fluid Flow in Microcirculatory Network and Interstitium in Tumors. Microvasc. Res. 1997, 53, 128–141.
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17.
- Zhang, K.; Fang, Y.; He, Y.; Yin, H.; Guan, X.; Pu, Y.; Zhou, B.; Yue, W.; Ren, W.; Du, D.; et al. Extravascular Gelation Shrinkage-Derived Internal Stress Enables Tumor Starvation Therapy with Suppressed Metastasis and Recurrence. Nat. Commun. 2019, 10, 5380.
- Roose, T.; Netti, P.A.; Munn, L.L.; Boucher, Y.; Jain, R.K. Solid Stress Generated by Spheroid Growth Estimated Using a Linear Poroelasticity Model☆. Microvasc. Res. 2003, 66, 204–212.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429.
- Milosevic, M.; Fyles, A.; Hedley, D.; Pintilie, M.; Levin, W.; Manchul, L.; Hill, R. Interstitial Fluid Pressure Predicts Survival in Patients with Cervix Cancer Independent of Clinical Prognostic Factors and Tumor Oxygen Measurements. Cancer Res. 2001, 61, 6400–6405.
- Jain, R.K. Transport of Molecules, Particles, and Cells in Solid Tumors. Annu. Rev. Biomed. Eng. 1999, 1, 241–263.
- Jones, D.; Wang, Z.; Chen, I.X.; Zhang, S.; Banerji, R.; Lei, P.-J.; Zhou, H.; Xiao, V.; Kwong, C.; Van Wijnbergen, J.W.M.; et al. Solid Stress Impairs Lymphocyte Infiltration into Lymph-Node Metastases. Nat. Biomed. Eng. 2021, 5, 1426–1436.
- D’Angelo, C.; Quarteroni, A. On the Coupling of 1d and 3d Diffusion-Reaction Equations: Application to Tissue Perfusion Problems. Math. Models Methods Appl. Sci. 2008, 18, 1481–1504.
- Huang, Y.L.; Tung, C.; Zheng, A.; Kim, B.J.; Wu, M. Interstitial Flows Promote Amoeboid over Mesenchymal Motility of Breast Cancer Cells Revealed by a Three Dimensional Microfluidic Model. Integr. Biol. 2015, 7, 1402–1411.
- Li, R.; Serrano, J.C.; Xing, H.; Lee, T.A.; Azizgolshani, H.; Zaman, M.; Kamm, R.D. Interstitial Flow Promotes Macrophage Polarization toward an M2 Phenotype. Mol. Biol. Cell 2018, 29, 1927–1940.
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248.
- Pisano, M.; Triacca, V.; Barbee, K.A.; Swartz, M.A. An in Vitro Model of the Tumor–Lymphatic Microenvironment with Simultaneous Transendothelial and Luminal Flows Reveals Mechanisms of Flow Enhanced Invasion. Integr. Biol. 2015, 7, 525–533.
- Carmeliet, P. Angiogenesis in Health and Disease. Nat. Med. 2003, 9, 653–660.
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92.
- Novikov, N.M.; Zolotaryova, S.Y.; Gautreau, A.M.; Denisov, E.V. Mutational Drivers of Cancer Cell Migration and Invasion. Br. J. Cancer 2021, 124, 102–114.
- Lusby, R.; Dunne, P.; Tiwari, V.K. Tumour Invasion and Dissemination. Biochem. Soc. Trans. 2022, 50, 1245–1257.
- Van Den Brink, A.; Suárez Peredo Rodríguez, M.F.; Foijer, F. Chromosomal Instability and Inflammation: A Catch-22 for Cancer Cells. Chromosome Res. 2023, 31, 19.
- ICGC Prostate UK Group; Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Högnäs, G.; et al. The Evolutionary History of Lethal Metastatic Prostate Cancer. Nature 2015, 520, 353–357.
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It Takes a Village. Nat. Rev. Cancer 2015, 15, 473–483.
- Clark, A.G.; Vignjevic, D.M. Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22.
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective Invasion in Breast Cancer Requires a Conserved Basal Epithelial Program. Cell 2013, 155, 1639–1651.
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114.
- Jorgens, D.M.; Inman, J.L.; Wojcik, M.; Robertson, C.; Palsdottir, H.; Tsai, W.-T.; Huang, H.; Bruni-Cardoso, A.; López, C.S.; Bissell, M.J.; et al. Deep Nuclear Invaginations Linked to Cytoskeletal Filaments: Integrated Bioimaging of Epithelial Cells in 3D Culture. J. Cell Sci. 2017, 130, 177–189.
- Harmati, M.; Gyukity-Sebestyen, E.; Dobra, G.; Janovak, L.; Dekany, I.; Saydam, O.; Hunyadi-Gulyas, E.; Nagy, I.; Farkas, A.; Pankotai, T.; et al. Small Extracellular Vesicles Convey the Stress-Induced Adaptive Responses of Melanoma Cells. Sci. Rep. 2019, 9, 15329.
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat. Cell Biol. 2019, 21, 9–17.
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848.
- Othman, N.; Jamal, R.; Abu, N. Cancer-Derived Exosomes as Effectors of Key Inflammation-Related Players. Front. Immunol. 2019, 10, 2103.
- Ghaemmaghami, A.B.; Mahjoubin-Tehran, M.; Movahedpour, A.; Morshedi, K.; Sheida, A.; Taghavi, S.P.; Mirzaei, H.; Hamblin, M.R. Role of Exosomes in Malignant Glioma: microRNAs and Proteins in Pathogenesis and Diagnosis. Cell Commun. Signal. 2020, 18, 120.
- Nahand, J.S.; Vandchali, N.R.; Darabi, H.; Doroudian, M.; Banafshe, H.R.; Moghoofei, M.; Babaei, F.; Salmaninejad, A.; Mirzaei, H. Exosomal microRNAs: Novel Players in Cervical Cancer. Epigenomics 2020, 12, 1651–1660.
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977.
- Wu, B.; Liu, D.-A.; Guan, L.; Myint, P.K.; Chin, L.; Dang, H.; Xu, Y.; Ren, J.; Li, T.; Yu, Z.; et al. Stiff Matrix Induces Exosome Secretion to Promote Tumour Growth. Nat. Cell Biol. 2023, 25, 415–424.
- Palomar-Alonso, N.; Lee, M.; Kim, M. Exosomes: Membrane-Associated Proteins, Challenges and Perspectives. Biochem. Biophys. Rep. 2024, 37, 101599.
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232.
- Hu, Q.; Su, H.; Li, J.; Lyon, C.; Tang, W.; Wan, M.; Hu, T.Y. Clinical Applications of Exosome Membrane Proteins. Precis. Clin. Med. 2020, 3, 54–66.
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human Colorectal Cancer Cells Induce T-Cell Death Through Release of Proapoptotic Microvesicles: Role in Immune Escape. Gastroenterology 2005, 128, 1796–1804.
- Shen, T.; Huang, Z.; Shi, C.; Pu, X.; Xu, X.; Wu, Z.; Ding, G.; Cao, L. Pancreatic Cancer-derived Exosomes Induce Apoptosis of T Lymphocytes through the P38 MAPK-mediated Endoplasmic Reticulum Stress. FASEB J. 2020, 34, 8442–8458.
- Zheng, D.; Huo, M.; Li, B.; Wang, W.; Piao, H.; Wang, Y.; Zhu, Z.; Li, D.; Wang, T.; Liu, K. The Role of Exosomes and Exosomal MicroRNA in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 8, 616161.
- Li, C.; Zhou, T.; Chen, J.; Li, R.; Chen, H.; Luo, S.; Chen, D.; Cai, C.; Li, W. The Role of Exosomal miRNAs in Cancer. J. Transl. Med. 2022, 20, 6.
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular Vesicles from Human Cardiac Progenitor Cells Inhibit Cardiomyocyte Apoptosis and Improve Cardiac Function after Myocardial Infarction. Cardiovasc. Res. 2014, 103, 530–541.
- Zhao, L.; Ma, X.; Yu, J. Exosomes and Organ-Specific Metastasis. Mol. Ther.-Methods Clin. Dev. 2021, 22, 133–147.
- Yang, X.; Zhang, Y.; Zhang, Y.; Zhang, S.; Qiu, L.; Zhuang, Z.; Wei, M.; Deng, X.; Wang, Z.; Han, J. The Key Role of Exosomes on the Pre-Metastatic Niche Formation in Tumors. Front. Mol. Biosci. 2021, 8, 703640.
- Bhatia, R.; Chang, J.; Munoz, J.L.; Walker, N.D. Forging New Therapeutic Targets: Efforts of Tumor Derived Exosomes to Prepare the Pre-Metastatic Niche for Cancer Cell Dissemination and Dormancy. Biomedicines 2023, 11, 1614.
- Wu, M.; Liang, Y.; Zhang, X. Changes in Pulmonary Microenvironment Aids Lung Metastasis of Breast Cancer. Front. Oncol. 2022, 12, 860932.
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-Derived Exosomes Induce N2 Polarization of Neutrophils to Promote Gastric Cancer Cell Migration. Mol. Cancer 2018, 17, 146.
- Rosell, A.; Martinod, K.; Mackman, N.; Thålin, C. Neutrophil Extracellular Traps and Cancer-Associated Thrombosis. Thromb. Res. 2022, 213, S35–S41.
- Morrissey, S.M.; Yan, J. Exosomal PD-L1: Roles in Tumor Progression and Immunotherapy. Trends Cancer 2020, 6, 550–558.
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of Pre-Metastatic Niche: The Landscape of Molecular and Cellular Pathways. Mol. Biomed. 2021, 2, 3.
- Haber, D.A.; Velculescu, V.E. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Discov. 2014, 4, 650–661.
- Pantel, K. Blood-Based Analysis of Circulating Cell-Free DNA and Tumor Cells for Early Cancer Detection. PLoS Med. 2016, 13, e1002205.
- Izumchenko, E.; Chang, X.; Brait, M.; Fertig, E.; Kagohara, L.T.; Bedi, A.; Marchionni, L.; Agrawal, N.; Ravi, R.; Jones, S.; et al. Targeted Sequencing Reveals Clonal Genetic Changes in the Progression of Early Lung Neoplasms and Paired Circulating DNA. Nat. Commun. 2015, 6, 8258.
- Birkenkamp-Demtröder, K.; Nordentoft, I.; Christensen, E.; Høyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbæk, M.; Jensen, J.B.; Ørntoft, T.F.; et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur. Urol. 2016, 70, 75–82.
- Spira, A.; Yurgelun, M.B.; Alexandrov, L.; Rao, A.; Bejar, R.; Polyak, K.; Giannakis, M.; Shilatifard, A.; Finn, O.J.; Dhodapkar, M.; et al. Precancer Atlas to Drive Precision Prevention Trials. Cancer Res. 2017, 77, 1510–1541.
- Gao, Q.; Zeng, Q.; Wang, Z.; Li, C.; Xu, Y.; Cui, P.; Zhu, X.; Lu, H.; Wang, G.; Cai, S.; et al. Circulating Cell-Free DNA for Cancer Early Detection. Innovation 2022, 3, 100259.
- Guerrero-Preston, R.; Valle, B.L.; Jedlicka, A.; Turaga, N.; Folawiyo, O.; Pirini, F.; Lawson, F.; Vergura, A.; Noordhuis, M.; Dziedzic, A.; et al. Molecular Triage of Premalignant Lesions in Liquid-Based Cervical Cytology and Circulating Cell-Free DNA from Urine, Using a Panel of Methylated Human Papilloma Virus and Host Genes. Cancer Prev. Res. 2016, 9, 915–924.
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated Digital Error Suppression for Improved Detection of Circulating Tumor DNA. Nat. Biotechnol. 2016, 34, 547–555.
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68.
- Fernandez-Cuesta, L.; Perdomo, S.; Avogbe, P.H.; Leblay, N.; Delhomme, T.M.; Gaborieau, V.; Abedi-Ardekani, B.; Chanudet, E.; Olivier, M.; Zaridze, D.; et al. Identification of Circulating Tumor DNA for the Early Detection of Small-Cell Lung Cancer. eBioMedicine 2016, 10, 117–123.

