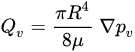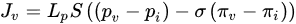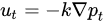1. Cancer Development
Cancer can be seen as a collection of diseases that evolve when the processes that regulate normal cell growth, cell division, and the entire life span become dysregulated. Consequently, the cells begin to proliferate out of control, prevent them from dying when they normally ought to, and cause the motility of other cells and tissues like blood vessels, immune cells, and various other types of normal cells to fuel the tumor’s growth advantage relative to the adjacent tissue. When the cancer progresses, specific cells inside the cancer take on specific modifications that offer them and their offspring cells the highest likelihood of growing and surviving. These alterations may involve the capacity to proliferate more rapidly, to manage to persist in the face of medical intervention, to infiltrate neighboring tissues and organs, to escape the body’s immune defense system, to enter the bloodstream and/or lymphatic system, to propagate without entering vascular systems, and to disseminate to remote areas of the organism. The majority of more advanced cancers display some, perhaps all, of these characteristics.
1.1. Driver Mutations Advance Cancerogenesis
Cancer leads to an imbalance of fundamental processes in the cell that subsequently cause alterations in DNA and in DNA-repair enzymes. As a consequence, cancer cells acquire multiple (epi)genetic changes in the course of their lifetime, and these changes occur one or two orders of magnitude more often compared to reproductive and normal somatic cells [
9]. Consequently, cancer cells collect numerous genetic changes during their lifespan, but only a limited number of them accelerate the progression of the cancer, the so-called driver mutations. Cancerogenesis has long been revealed as a multistage disease. Driver mutations can differ according to the cancer type and the individual patient, and their impact on cancer may be time-delayed or immediate. For example, they can stay dormant for quite some time and turn into drivers only at specific phases of cancer, or they can promote oncogenesis only in combination with additional mutations. The large mutational, biochemical, and histological heterogeneity of tumors renders the identification of driver mutations very difficult. One of the first findings was that the mortality rate for certain types of cancer increases with the sixth power of the patient’s age. A numerical model has been developed that predicts a number of successive driver mutations and different cancer stages [
10]. Additional investigations identified a limited number of mutations that accelerate the development of cancer (driver mutations) [
11,
12]. For example, roughly a single driver mutation for each patient was detected in sarcoma, thyroid, and testicular cancer, and approximately four driver mutations for each patient with bladder, colorectal, and endometrial cancer [
13]. Nevertheless, it is generally agreed that most mutations in cancer are broadly neutral, which are referred to as passenger mutations, and do not encourage the pathogenesis of cancer. The overwhelming majority of driver mutations are either replacements of single nucleotides or point mutations.
Apart from genomic mutations, epigenetics concerns changes in DNA that are not associated with a modification of the DNA base sequence. The epigenetically altered genome is termed the “epigenome”. An important epigenetic process involves the packaging of a cell’s DNA within a cell nucleus. The latest findings strengthen the idea that mutations in the epigenetic signal transduction apparatus, comprising histones, such as K27M mutations in histone H3 in gliomas [
14], and chromatin remodelers, such as in the genes ARID1A and ARID1B [
15], are prospective new epigenetic markers in cancer [
16].
1.2. Establishment of Basic Primary Cancer Models
Cancer is evoked by a broad range of factors, including chemical carcinogenesis, epigenetic changes, somatic mutations, and viral infections [
17,
18]. The way normal cells turn into cancer cells is currently based on two different models (
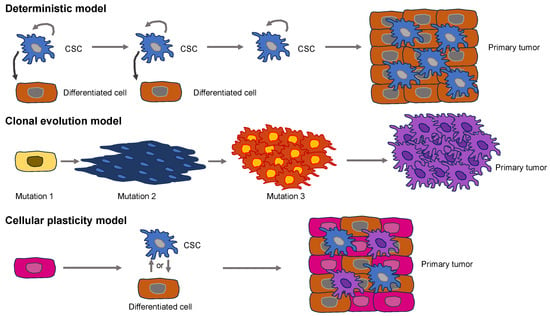
Figure 1). Supporters of the deterministic model propose the hypothesis that somatic stem cell transformation by somatic mutations produces a specific subset of cancer cells capable of self-renewal, which are known as cancer stem cells (CSCs) [
19,
20]. CSCs generate subsidiary cells that have a restricted tumorigenic and metastatic capacity and subsequently develop primary tumors [
21,
22].
Figure 1. Models for the initiation of primary solid tumors. The deterministic model (upper images) presents CSCs at the apex of the hierarchical organization. CSCs possess the potential for self-renewal and can produce differentiated cells that are less cancerogenic. The clonal evolution model (middle images) accumulates genomic mutations or epigenetic alterations that enable cancer cells to proliferate faster compared to normal, healthy cells. The cellular plasticity model (lower images) postulates the idea that CSCs do not represent cells of origin. Instead, plasticity occurs in CSCs, or differentiated cells, that finally culminate in the primary tumor.
The model of clonal evolution of carcinogenesis, which is commonly referred to as the stochastic model, implies that mutations or epigenetic changes bestow selective reproductive predominance on a cell over healthy cells, resulting in unrestrained growth and a primary solid tumor [
23,
24]. The clonal evolution model is similar to a Darwinian natural selection model, which is an adaptive system. The model of clonal evolution predicts that selection at various phases of tumor growth leads to enhanced genetic and epigenetic changes and a reduction in tumor suppressive defense mechanisms, resulting in susceptibility to oncogenesis [
25]. None of these models accounts for the high level of heterogeneity present in primary solid tumors [
26]. Recently, a fusion of these two models, referred to as the cellular plasticity model, has been advocated (
Figure 1). It assumes that the “original cell” in this case is different from a CSC. Rather, the cellular plasticity model assumes that healthy cells are naturally plastic and may exhibit phenotypic alterations upon being subjected to a stimulus, either internally or externally [
27]. This congenital plasticity allows healthy cells to evolve by undergoing epigenetic and phenotypic modifications, enabling transformation into CSCs. Exterior impulses are capable of causing the buildup of multiple mutations throughout the cancer cells, leading to a huge heterogeneity within primary solid tumors [
17]. A heterogeneous solid tumor arises from both kinds of mutated cells [
28].
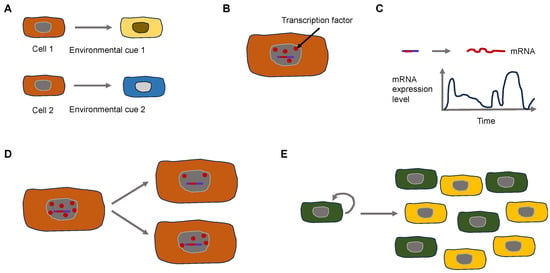
Why does phenotypic heterogeneity arise on a mechanistic basis? The expression of genes constitutes a stochastic or “noisy” biological process. This noise can arise in two different types. It can be broken down broadly into two principal categories: Firstly, isogenic cells/individuals that obtain the same pieces of information from the surrounding environment can result in varying expressions of a phenotypic characteristic. Isogenic cells vary because of the noise generated through the process of splitting the cell components binomially during the moment of cell division [
29,
30]. Consequently, the inherent stochasticity of biochemical processes like transcription and translation produces “intrinsic” noise. Secondly, isogenic cells/individuals that obtain varying pieces of information from the surrounding environment can result in varying expressions of a phenotypic characteristic. Fluctuations in the abundance or states of other cellular compounds elicited by the surrounding environment can indirectly result in fluctuations in the expression of a certain gene and therefore generate “extrinsic” noise [
31] (
Figure 2).
Figure 2. Extrinsic and intrinsic signals control the heterogeneity of cancer cells. (A) Extrinsic cues can lead to cell heterogeneity. Two isogenic cells are subject to different environmental cues due to broad microenvironmental variations that evoke various responses. (B–E) Intercellular variations lead to heterogeneity due to intracellular variations. (B) A small number of transcription factors (red dots) require different times to bind to the promotor region (short red line) on the DNA (blue line). (C) The activity of cellular processes such as transcription is time-dependent. (D) Non-equal cell division can cause intercellular differences. (E) Feedback circuits in cellular populations result in all (green) or none states (yellow) in cellular physiology.
Does a universal mechanical phenotype of cancer cells exist in general or at specific time points of tumorigenesis and/or malignant progression? The findings of the physical oncology research community point towards the fact that the relationship between the biophysical TME and genetic modification exerts a considerable influence on the progression of tumors. Cancer cells in particular and the stromal cells connected to them modify their individual cytoskeletal and physical characteristics while also restructuring the surrounding microenvironment with abnormal mechanical characteristics [
5]. Finally, these altered mechano-omics of cancer tissues and their components profoundly displace the mechanotransduction paradigms in cancer and stromal cells and activate oncogenic cues inside the neoplastic niche to promote cancer progression.
Although there are many possibilities for tumor cell diversity, is there still a possibility that a certain cell phenotype, such as the mechanophenotype, is not different but the same? Moreover, is this mechanophenotype found universally in the different tumors, does it also exist during malignant progression, or is it at least also universally changed?
2. Universal Properties of Cancer and Their Effect on Cancer Cell Function
The mechanical phenotype can be characterized by considering the cell as a material. It is a pivotal factor as it dictates the interaction between forces such as tension, pressure, and entrapment and the consequent alterations in cell morphology, such as cell shape and physical size. The mechanical phenotype can be expressed by various measurements like viscoelasticity, cell deformability, cell adhesion characteristics, and cell shape. To address the question of whether there is a universal mechanical phenotype of cells, it is possible to consider the function of certain proteins, such as Caveolin1 (Cav1). Specifically, the focus is on emphasizing the involvement of caveolae and caveolar constituents, particularly Cav1, in the process of incorporating the mechanical forces transferred by the ECM [
32] and the specific involvement of cellular cytoskeletal remodulation as a mechanism of action whereby caveolae and caveolins can react to and remodel the ECM [
33,
34,
35].
In general, it is the cellular mechanical phenotype that determines survival after deformations caused by entrapment and liquid streaming. The concept involves the assumption that cancer cells are sculptural (plastic) and acquire various mechanical phenotypes when exposed to various geometries, which promote their survival. Therefore, an attractive objective of physical cancer research is to interfere with the capacity of cancer cells to take on different mechanical states. For this reason, the mechanosensory process of cancer cells must be investigated. The ability of cells to recognize stiffness is necessary to react to the rigidity of the matrix. It was investigated how healthy cells and cancer cells exhibit discrete mechanical characteristics [
36]. Cancer cells appeared softer compared to their healthy control equivalents, irrespective of the specific cancer type, such as breast, bladder, cervix, ovarian, pancreatic, or Ha-RasV12-transformed cells [
37]. The measurements were conducted with atomic force microscopy (AFM). When growing on ECM matrices of different stiffness, low stiffness can impair the proliferation of healthy cells [
38], whereas this effect is not attained by cancer cells and transformed cells. Hence, cancer cells experience a modification of their mechanical phenotype, which entails a softening of the cell and a reduction in the sensation of stiffness. In a groundbreaking investigation, it was shown that mesenchymal stem cells (MSCs) alter their morphology and develop towards lineage-specific differentiation when grown on matrices that exhibit varying, physiologically important matrix stiffnesses [
39]. Cancer cells are capable of undergoing malignant transformation and taking on a mesenchymal phenotype.
Cav1 acts as a small oligomeric scaffold protein that is generally necessary for the development of membrane curvatures in structures like caveolae [
40,
41]. In addition, Cav1 connects to multiple other proteins, regulates the homeostasis of cholesterol, and controls a multitude of cell processes, including endocytosis, internalization of receptors, accumulation of cholesterol, cell signal transduction pathways, proliferation, and cell death [
41,
42]. Cav1-driven regulation of the signaling cues is critical in cancer. An example of this is the crosstalk of Cav1 and Rho GTPases, including RhoC, that promotes the emergence of metastases through inducing the expression of α5 integrin and the Src kinase-facilitated activation of the p130Cas/Rac1, FAK/Pyk2, and Ras/Erk1/2 signal transduction pathways [
43,
44]. It is important to note nonetheless that Cav1 also acts as a tumor suppressor by helping E-cadherin in the sequestration of β-catenin, thereby impairing the β-catenin/Tcf-Lef-dependent transcriptional activation of genes encompassing survivin, cyclooxygenase-2, cyclin D1, and multiple other proteins fostering the development of cancer [
45,
46]. Therefore, Cav1 fulfills functions of opposite outcomes, such as tumor suppressor and promotor in cancer, which have been reviewed in [
41,
47,
48].
Cav1 suppresses tumor formation via regulation of contractile tension in epithelia [
49]. In specific detail, caveolin’s contractile tension control is required to remove oncogene-transfected cells through the process of apical extrusion. Lack of caveolin-1 enhanced steady-state tensile stresses within epithelial monolayers, and consequently, the lack of Cav1 in the epithelial cells encircling oncogene-expressing cells hampered their apical extrusion.
Contractile force at the adherent junctions (AJs) mirrors the action of the actomyosin cortex, which is linked to E-cadherin-driven cell-cell adherence [
50]. To comprehend how caveolae modulate junctional tension, first the actomyosin cortex at AJs was analyzed. Unexpectedly, the concentrations of non-muscle myosin II and phosphorylated myosin regulatory light chain (pMLC) concentrations at the junctions of Cav1-KD cells could not be determined even though mechanical tension was elevated. This suggests that although myosin II is required for tension, alterations in this motor cannot straightforwardly account for the elevation of tension within Cav1-KD cells. Neither the expression of E-cadherin nor the dynamics of the junctions were changed. In confocal microscopy, the F-actin values were slightly but persistently increased. This indicates that an actin modulatory signaling mechanism could be in charge of the junctional tension enhancement in Cav1 KD.
To this end, the architecture of the F-actin network was primarily characterized using structured illumination microscopy (SIM). Apical junctional F-actin occurred with greater condensation in Cav1-KD cells, and fewer superimposed filaments and bundles were visible after the skeletonization of the microscopic images. Thus, the junctional cytoskeletal organization was modified through the knockdown of Cav1. This assumption was confirmed based on the measurement of the nematic order of F-actin at the junctions following the Fourier transformation of the fluorescence signals obtained from SIM images [
51]. The nematic order coefficient was decreased in Cav1-KD cells, which is in line with a stronger co-linear or bundled arrangement of actin filaments. Ultimately, actin dynamics were characterized based on the expression of G-actin labeled with photoactivatable GFP (PAGFP-G-actin), and its fluorescence loss after photoactivation (FLAP) was assessed at the junctions. The T
1/2 half-life of fluorescence decay was markedly elevated at the junctions of Cav1-KD and Cavin1-KD cells relative to controls, indicating that the F-actin pool remained more stable. Altogether, these results imply a mechanism that functions to stabilize F-actin and facilitate its bundling at AJs to be hyperactive in Cav1 KD cells. Formins seemed to be appealing contenders for imparting these effects, as they facilitate the assembly and stabilization of non-branched F-actin structures and operate at cell-cell junctions [
52,
53,
54]. Similarly, the broad-spectrum formin inhibitor SMIFH2 applied to Cav1-KD cells restored F-actin levels at the junctions and normalized F-actin architecture and dynamics. It is important to emphasize that SMIFH2 compensated for the increased junctional tension observed in Cav1-KD cells. This suggests that an excessively active formin is likely in charge of the elevation of junctional tension when Cav1 is knocked down. Caveolae have lately been identified as mechanically active organelles that contribute to the mechanical damage protection of tissues, such as mechanical stress [
55,
56]. An understanding of this protective mechanism emerged from the finding that the caveolae become flattened when membrane tension rises. It was suggested that a membrane reservoir is thereby freed to buffer the changing membrane tension in a passive manner [
57,
58,
59,
60]. These results highlight another way in which caveolae can impact the mechanics of epithelial monolayers. In this case, the removal of caveolae enhances cortical contractility through stimulation of a phospholipid signal transduction pathway that aims at the actin cytoskeleton. It is proposed that caveolae modify active tissue tension by limiting the activity of this signaling mechanism. In this way, caveolae contribute to establishing an acceptable mode of epithelial tension for oncogenic cells that can be eradicated through apical extrusion. Cav1, known to be suppressed in a variety of cancer cells and oncogene-transformed cells, controls the mechanical phenotype [
61,
62]. Cav1-driven elevation of RhoA activity and Y397FAK phosphorylation guided actin cap production, which was positively associated with cell elasticity and stiffness perception in fibroblasts. Ha-RasV12-induced cell transformation and alterations in mechanical phenotype can be reverted by re-expression of Cav1 and mimicked when Cav1 is silenced in normal fibroblasts. Finally, this study revealed a novel function of Cav1 and identified a connection between mechanical phenotype and the transformation of cells. Consequently, mechanical properties can also be used as indicators of cell transformation. In summary, the role of Cav1 in cancer, in particular the comprehension of the canonical (Cav1 located in the plasma membrane) and non-canonical pathways (Cav1 situated in organelles and exosomes), is connected to the protein’s double function as tumor suppressor and facilitator of metastasis [
63].
The general heterogeneity of tumors argues against the universal nature of the characteristics of cancer cells. Cell shape heterogeneity was found to be more tightly coupled to the mechanical state of the cells. However, single cells in multicellular spheroids display a lower level of mechanical heterogeneity than individual cells grown in monodisperse 3D culture systems. The reduced heterogeneity among cells found in spheroids implies that there is mechanical cooperation among the cells that comprise a solitary spheroid. Another possibility for the reduced heterogeneity within multicellular spheroids lies in their rather simple composition and architecture. There are also more intricate multicellular models available, such as organoids or even better tumoroids, which means tumor-like organoids. Organoids comprise three-dimensional complex ex vivo tissue cultures that can be obtained from embryonic stem cells, induced pluripotent stem cells, or tissue-resident progenitor cells. They have spatially limited lineage connectivity and higher-order self-organization, which renders them attractive quasi-physiological models [
64]. Organoids still lack the full composition of cells, molecules, and factors that reside within a patient’s solid tumor. Although organoid cultures are superior to spheroid culture models, they still have weaknesses. Therefore, it is advantageous to generate samples directly from freshly isolated (resected or biopsy) patients’ cancer tissue and to preserve the complete structural organization of the TME and the ECM [
65].
Organoids provide a biologically meaningful stage for enhancing translatability. Co-cultures represent no novel concept in the experimental work, as they are frequently utilized to investigate interferences between epithelial cells and other relevant cell populations, for example, lymphocytes, neurons, and blood vessels, as recently reviewed in [
66]. The cultivation of epithelial cancer organoids with immune cells has provided valuable findings on the pathogenesis of various cancers, and the opportunity to genetically engineer these types of organoids in the absence or presence of immune cells offers a distinct and pertinent model for the examination of carcinogenesis [
67,
68,
69]. The co-culture of mouse tumor organoids and adipocytes delivered new findings on colorectal cancer. For example, it has been demonstrated that adipocytes stimulate the proliferation and dedifferentiation, which is evidenced by elevated Lgr5 and CD44 and reduced mucin-2 and sucrase-isomaltase mRNA expression levels, of colon cancer organoids [
70]. The researchers also hypothesize that adipocytes act as a metabolic regulator and energy supplier to support the growth of colon cancer cells, which is a candidate mechanism to account for the association between obesity and colon cancer. The ECM is not a mere passive spectator in cancer biology; nevertheless, the biological implications are frequently not investigated or considered in conventional laboratory experiments [
71]. Co-culture experiments can solve this problem. Established organoids of pancreatic ductal adenocarcinoma, for instance, normally form ductal and basement membrane architectures, but this structure is destroyed upon co-culture with pancreatic stellate cells within a collagen matrix, resulting in deterioration of the basement membrane and enhanced invasion of the collagen matrix [
72]. In addition, co-cultivation of pancreatic cancer organoids together with both stromal and immune cells results in the activation of myofibroblast-like cancer-associated fibroblasts, a phenomenon that was not evident in 2D cell culture models [
73]. A model system that enables the interplay between cancer cells, stromal cells, and immune cells is consequently crucial for examining the pathogenesis of cancer.
New techniques are constantly being developed to optimize organoid cultures, such as the incorporation of self-generating hydrogels consisting of an ECM extracted from human tissue in place of mouse Matrigel. For instance, a methodology has been developed for preparing extracts from the ECM of breast mammary glands that can undergo spontaneous gelling and produce hydrogels [
74]. The important point is that these hydrogels sustain biological signaling reactions that differ between cancer and normal epithelial organoid cultures [
74]. Culture systems with air-liquid boundaries, in which the basal surface of the stem cells is in direct physical exposure to the culture medium and the apical surface is in contact with air, are equally interesting. This arrangement can more precisely reproduce the characteristics of the TME in specific types of cancer, like the luminal surface of colorectal carcinoma [
75].
Based on the issues discussed, the question can be asked whether it is likely that cancer cells have the same mechanical properties from patient to patient, regardless of the type of cancer and the variables. However, the stage of cancer seems to have an influence on the mechanical phenotype, as there appears to be a strong correlation. The majority of research carried out to date has shown that individual cancer cells are softer compared to healthy cells, as illustrated in several review articles [
76,
77,
78], which all focus on AFM stiffness analysis. The differences in the stiffness of cancer cells with varying invasive abilities, however, are subject to less agreement.
Recently, it has been revealed that mechanical characteristics at the level of the cell (stiffness, viscoelasticity) and at the level of the plasma membrane (fluidity) are interlinked [
79]. More invasive cancer cells have been shown to either soften with magnetic tweezers with fibronectin-coated superparamagnetic beads, such as ovarian cancer cell lines [
80] and with AFM, such as ovarian HEY, HEY A8, OVCAR-3, and OVCAR-4 cancer cells [
81] and B16 melanoma cell variants [
82], or stiffen, such as prostate, liver, and breast cancer cell lines [
83,
84,
85,
86], during the course of cancer advancement. Moreover, even when breast cancer cells are measured in an adherent and non-adherent state using AFM, there is still the finding that the softer breast cancer cells are more invasive into 3D collagen matrices and cause more fiber displacement when invading these 3D collagen fiber scaffolds [
87]. Some of the observed inconsistencies may be attributable to the considerable diversity of cancer cells and the signaling pathways participating in the process of invasion [
88]. Within this, higher stiffness (apparent elastic modulus and E
0) was observed in cells with a mesenchymal phenotype and the highest migration performance (SW480), and lower stiffness in cells with an epithelial phenotype and low migration performance (HT29) [
79]. Cell height and power-law exponent were increased in the softer HT29 cells, which is consistent with earlier findings [
89,
90]. The Newtonian viscosity coefficient η of the utilized viscoelastic model exhibited a positive correlation with stiffness. This result is not entirely clear, as it could be related to the viscosity of the cytoplasm in the vicinity of the cortex, which is also investigated at the penetration depths used (800–1500 nm, typically) [
79]. However, there is currently no further data to substantiate this conclusion, and further research is needed to make a statement [
79].
3. Malignant Cancer Progression (Cancer Metastasis)
Cancer that has disseminated to other areas of the body, which is commonly referred to as metastatic disease, is the leading contributor to the majority of cancer deaths. Cancer cells surmount numerous hurdles, such as surveillance by the immune system, to propagate to secondary sites effectively [
91]. A plethora of research performed over the last two decades heavily implies that mechanical forces are also implicated in cancer progression and responses to traditional therapeutic regimens [
92,
93,
94]. These forces also include fluid mechanics, which are increasingly coming into focus. On their route to establishing a metastasis, cancer cells and factors released by the cancer utilize and harness three key body fluids: the blood, lymph, and interstitial fluid [
95,
96,
97]. The important fact is that fewer than 0.01% of the thousands of cancer cells penetrate the bloodstream and survive to develop metastases [
98,
99]. Thus, it is not likely that all cancer cells can be cleared from the patient by surgery. Thus, it is critical to comprehend the specific steps of the metastatic cascade to develop inhibitory treatments. All of these steps are concerned with mechanical encounters between the cancer cells and the various microenvironments they experience during metastasis [
95]. Circulating tumor cells (CTCs) and their derived products, comprising soluble factors, cell-free DNA, and extracellular vesicles (EVs), can migrate directly through the hematogenous circulation [
91,
100] or sequentially exploit both the lymphatic and vascular systems to populate distant organs [
101,
102,
103].
This idea that fluid mechanics can influence metastasis goes back to an early investigation that pioneered the “hemodynamic theory” and demonstrated that arterial blood flow in specific organs is positively correlated with the occurrence and patterns of metastasis [
104], which demonstrates a connection between fluid mechanics and the secondary location of metastasis.
During transportation in liquids, CTCs are exposed to and act on different mechanical forces, which can affect their destiny in a variety of ways. For example, high shear forces acting on CTCs can trigger mechanical stress that results in cell fracture and death [
105], while intermediate shear forces have been demonstrated to promote the intravascular dormancy phase and the extravasation of CTCs [
106]. A better understanding of the mechanical forces to which CTCs and tumor-associated material are subjected in fluids is therefore critical to completely unraveling the metastatic cascade and defining susceptible CTC stages for therapeutic interference. This investigation reveals a novel mechanism by which a VEGF-VEGFR2-AKT-ATOH8 signaling axis induced through cyclic laminar shear stress (LSS) confers survival to mimic circulating tumor cells (m-CTCs) [
107].
3.1. Increased Vascular Permeability around Cancers
The effect of enhanced permeability and retention (EPR) involves the extravasation of blood components from leaking tumor-induced vessels and their retention in the TME, thereby increasing interstitial pressure. Cancer cells can easily enter the leaky vessels to spread to targeted tissues and organs. As expected, these suspended cells with a CK8
+/CD45
−/DAPI
+ phenotype have been seen in blood vessels and are referred to as m-CTCs. Quantitative polymerase chain reaction, western blotting, and immunofluorescence were employed to assess the alterations in gene expression of m-CTCs that exhibit sensitivity to LSS pacing. In addition, the expression of atonal bHLH transcription factor 8 (ATOH8) in CTCs of 156 CRC patients and mice was investigated using fluorescence in situ hybridization and flow cytometry [
107]. The m-CTCs actively reacted to LSS by inducing the expression of ATOH8, which is a fluid mechanosensor involved in intravascular surveillance and the plasticity of metabolism. Notably, ATOH8 was observed to be upregulated through activation of the VEGFR2/AKT signal transduction pathway facilitated through LSS-triggered VEGF secretion. ATOH8 subsequently transcriptionally induced HK2-driven glycolysis, thereby enhancing the intravascular survival of colorectal cancer cells within the blood circulation.
The onset of fluid mechanics was the detection of diffusive transport of oxygen around blood capillaries, which was reviewed in [
108]. Pioneering contributions have been identified that advance the application of fluid mechanics concepts to accompany genomic and molecular signaling investigations for cancer research [
109,
110,
111,
112,
113]. The fluid mechanics of the cancer microenvironment were also explored in the work by Maeda and colleagues [
114]. With the concept of the enhanced permeability and retention (EPR) effect, Maeda and colleagues [
114] advocated the critical contribution of fluids extravasating from the tumor vasculature leading to elevated interstitial pressure, which constitutes a dominant element in the TME and a governing mechanism for cancer treatment.
3.2. Bodily Fluids
Blood, lymph, and interstitial fluid properties can be characterized based on their biophysical attributes, which are affected by their unique constitution and features, providing an appreciation of the mechanical stress that each fluid can exert on CTCs and other tumor-derived constituents [
115,
116]. These interconnected partitions exhibit various flow modes (which rely on the magnitude of the dimensionless Reynolds number, among other factors) and flow velocities that are encountered or utilized by the CTCs and/or tumor-secreted matter during transport. In the lymphatic system, for example, it is a generally laminar, pulsating flow with low amplitude, which is primarily powered by viscosity and exhibits low velocities [
117]. Conversely, blood possesses a far higher density of circulating particles (blood cells and other factors) and has faster flow rates because of the heart’s pumping capacity. Moreover, the blood flow in arteries can be pulsating with high amplitude and turbulent flow, while the flow in veins is generally laminar [
118]. In addition, the biophysical cues vary based on the flow and type of vessel, as well as the nature of the organ. In total, CTCs and tumor-derived material moving in the circulatory system are subjected to shear rates ranging from around 10 s
−1 in the lymph [
117] to around 1000 s
−1 in large arteries [
118,
119].
Blood and lymph play a part in the interstitial fluid flow that typifies cancer [
94]. The interstitial fluid produced due to high capillary blood pressure and cellular pressure within the solid primary tumor [
120] is dissipated through the lymphatic system and its primary valves, which are under lower pressure. This directional shift was characterized in a model [
121] on the principle of Darcy’s law and is primarily related to the variations in pressure, surface area, and hydraulic conductivity among these networks. The interstitial fluid pressure (IFP), intriguingly, not only eases the spreading of cancer cells and tumor-associated material [
96], but also has key implications for the infiltration of drugs into the primary cancer site [
94].
3.3. Tumor Interstitial Fluid Facilitates the Cancer Cell’s Migration and Invasion
Solid primary cancers create a complex microenvironment comprising cancer cells, stromal cells, the ECM, blood vessels, and lymphatic vessels. When cancers grow to 1–2 mm, they need to become vascularized to elevate oxygen levels, and hence they need to recruit nearby blood vessels or induce the growth of new blood vessels [
122]. The growth of the primary solid tumor depends on angiogenesis, which offers oxygen and required nutrients through the de novo formation of new blood vessels, and lymphangiogenesis, which functions in the elimination of excessive fluids and the dehydration of cancer cells and tumor-secreted factors released from the tumor [
123]. Moreover, both fluid systems enable the transportation of immune cells into and out of the tumor, and the lymphatic circulatory system directs local immune surveillance through the oversight of adjacent lymph nodes [
124].
The Darcy model accounts for variations in IFP inside the tumor that promote the diffusion and convection of fluids from the blood into the outflowing lymphatic system [
96]. In primary tumors, the IFP gradient is exacerbated by the incompleteness of the immature tumor-associated blood vessel system, which contains many “holes” [
125]. Moreover, fast tumor growth leads to massive solid stress, which subjects the tumor-associated vascular network to additional vasoconstriction due to compression and tension [
126,
127]. Thereby, a high IFP is produced inside the tumor tissue. In an autochthonous mouse model for pancreatic ductal adenocarcinoma, the intratumoral IFP was over nine times higher compared to the IFP found in equivalent healthy tissue [
128,
129]. Even though a high IFP value is evident in the core of the tumor [
128], the IFP value decreases at the tumor periphery, resulting in an interstitial fluid flow across the peritumoral stroma into the lymphatic vessels [
96]. The interstitial fluid flow consists of both convection and diffusion fluxes in the direction of the periphery [
130], with velocities of about 0.001–0.004 mm s
−1 [
131]. At the same time, the stress puts pressure on the blood and lymph vessels [
132], which impedes the oxygen supply and homeostasis of the solid tumor. Conversely, cancer cells are prone to enhance the liberation of pro-angiogenic factors [
120], resulting in an abnormal and hyperpermeable blood circulation (low velocity and very heterogeneous), which further promotes the malignant response of cancer cells.
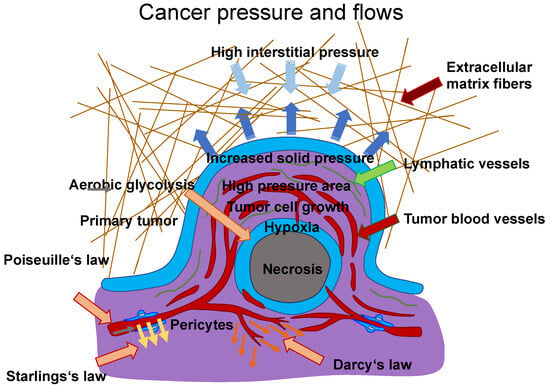
A typical case of vascular flow and transport is presented in, in which scientists explain a conceptual approach for incorporating a discrete 1D model of tissue vasculature into a 3D continuum model of interstitial trafficking [
133]. Fluid shear flow across a meshwork of vessels is characterized by Poiseuille’s law, which links blood flow to channel radius, pressure, and the viscosity of the blood liquid. The transfer through vessel walls can be characterized by Starling’s law, which links the rate of extravasation to vessel permeability and the pressure mismatch between the vessel and the tissue. The speed and direction of the interstitial flux can be determined by Darcy’s law, which connects these variables with the pressure gradient and the tissue’s hydraulic conductivity. To maintain the unique architectural structure of the vascular system, an approach associated with a continuous 3D model of the interstitial space that is not limited to a spatially averaged variable was established. These three essential relations are illustrated in
Figure 3, which are employed to determine the characterization of intravascular and interstitial flow. Poiseuille’s and Starling’s laws are integrated into this drawing (
Figure 3). Poiseuille’s law (Equation (1)) inks the intravascular flux
𝑄𝑣(Figure 3, gray arrow) to the radium of the vessel R, the dynamic blood fluid viscosity µ, and the intravascular pressure gradient 𝑝𝑣. Starling’s law (Equation (2)) links the rate of extravasation 𝐽𝑣 (Figure 3, yellow arrows) to the hydraulic conductivity of the vascular wall 𝐿𝑝, the vascular surface area S, the reflection coefficient σ, the vascular oncotic pressure 𝜋𝑣, and the interstitial oncotic pressure 𝜋𝑖. Darcy’s law (Equation (3)) connects the interstitial flow speed 𝑢𝑡 (Figure 3, orange arrows) to the interstitial tissue hydraulic conductivity 𝑘 and the interstitial pressure gradient 𝑝𝑡. These three linkages can be found within the overall published literature on the physical modeling of tumor-based vascular flux and angiogenesis.
Figure 3. Model of flows between blood vessels and tissues. The Poiseuille (grey arrow), Starling (yellow arrows), and Darcy (orange arrows) laws can describe the flux within blood vessels and tissues. The extravasation flow can be characterized by Starling’s law, which takes into account the pressure on both faces of the capillary, such as vascular (arterial or venous) and interstitial pressure. The overall hydrodynamics can be described using hydraulic conductivity coefficients for the arterial and venous flows to determine the singular performance of the vascular resistance. Darcy’s law can be used as a model for the movement of cancer cells with both inhomogeneous and isotropic conductivity. Therein, the cancer cells are treated as a fluid with constant density. This “cancer cell fluid” flows through a porous environment, such as the ECM, which is defined as rigid and immobile. Thus, the porosity is constant. Simulations have shown that the tumor mass increases from regions with high conductivity to regions with low conductivity if the tumor form is not altered. Thus, Darcy’s law states that a higher flow velocity is present in areas exhibiting higher conductivity. Poiseuille’s law states that the flow is highest at the vessel wall due to the enhanced fluid shear stress, which has an impact on cancer cell survival within vessels. In addition, there is high interstitial pressure (light blue arrows) from the TME, which compresses the tumor mass. The dark blue arrows indicate the expansion pressure caused by the proliferating tumor mass in the direction of surrounding tissue. Hypoxic conditions prevail in the core of a tumor mass due to fewer blood vessels, and some tumor masses are necrotic in their core area. Aerobic glycolysis, which requires oxygen, is one of the hallmarks of cancer and can only take place where sufficient oxygen is available.
Poiseuille’s law is provided in the following Equation (1):
Darcy’s law is given in Equation (3) as follows:
The interstitial fluid of cancer cells promotes the migration and invasion of cancer cells. The interstitial convection flux holds the capability to accelerate the invasion of glioblastoma cells and to promote the migration of amoeboid human breast carcinoma cells towards the lymphatic drainage system, where cancer cells can exit the primary tumor with the support of macrophages [
134]. Interstitial fluid flow alters stromal cells by enhancing the polarization of macrophages, which encourages the migration of cancer cells [
135]. In a mouse model of breast cancer, migrating macrophages are transformed into sessile perivascular macrophages [
136]. However, the effect of the interstitial flux is still elusive. As collagen I stimulates mesenchymal motility, cancer cells may migrate in the opposite direction of convective flux. The flux of interstitial fluid interacts with the flux of luminal vascular tissue to promote the intravasation of cancer cells [
137]. The motion of the interstitial fluid affects the migration of cancer cells toward the lymphatic and blood vessels. It has been hypothesized that convective pressures associated with lymphatic and blood circulation push tumor-related contents and cells to aid their targeted spread (as well as the spread of soluble melanoma factors, or EVs), toward the vascular events or ECM at the circumference of tumors. There is not enough scientific substantiation for this hypothesis. Other mechanisms, like postnatal angiogenesis (the formation of new blood vessels through recruited endothelial progenitor cells from the bone marrow) and vasculogenic mimicry, are possibly accountable for the formation of blood vessels [
138].
4. Chromosomal Instability, Exosomes, and Cell-Free DNA Foster Cancer Metastasis
Cancer metastasis is the hallmark of cancer, accounting for the majority of cancer-related deaths. However, it is still not clearly revealed. Invasion marks the first point in the metastatic cascade, when cancer cells gain the capacity to migrate, invade nearby tissue, and penetrate lymphatic and blood vessels to spread. A contributing part of genetic alterations in invasion is not generally agreed upon. The skeptical argument is that cell motility depends exclusively on external stimuli like hypoxia, chemoattractants, and mechanical factors such as the stiffness and/or viscoelasticity of the ECM. In tumor hypoxia, the content of oxygen is lowered from 4.6% to 9.5% in healthy tissues to less than 1–2% [
139]. However, there is growing evidence that mutations can initiate and enhance the migration and invasion of various types of cancer cells. The published literature on the implications of chromosomal instability and genetic mutations on the migration and invasion of cancer cells is presented in a recent review [
140]. Chromosomal instability, release of exosomes, and cell-free DNA (cfDNA) of primary tumors, normal cells, or cancer cells have emerged as hallmarks for the malignant progression of cancers. All of these features appear to be triggered by mechanical influences such as interstitial flow, external forces, and/or stiffness. As cancer cells are subject to mechanical cues, chromosomal instability may enable cancer cells to adjust to different environmental conditions by potentially adjusting their mechanophenotype and may help them to disengage from the primary tumor and subsequently metastasize. The stiffness-driven release of exosomes can reduce the response of immune cells by provoking their apoptosis. The cfDNA, which can also be released from exosomes, can protect cancer cells in the vascular system from destruction by the immune response, as the specific receptors existing on the cancer cells cannot be identified. Finally, these events can all contribute to cancer metastasis.
4.1. Chromosomal Instability
Cancer cells start to spread from the primary tumor before the first steps of the invasion-metastasis cascade take place [
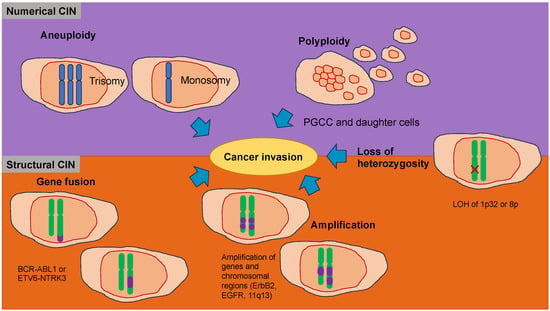
141]. The metastatic cascade is the result of chromosomal instability due to ongoing errors in the separation of chromosomes at the time of mitosis (
Figure 4). Errors in chromosome segregation lead to disruption of micronuclei and release of genomic DNA into the cytosol, which consequently activates cytosolic DNA-sensing signaling pathways (cyclic GMP-AMP synthase stimulator of interferon (IFN) genes) and subsequent nuclear factor-κ-light-chain-enhancer of activated B (NF-κB) signaling downstream pathways [
142]. Research indicates that the type of primary seeding cancer cell dictates the varying metastatic characteristics in terms of growth and responsiveness to treatment [
143,
144]. In vivo and in vitro trials demonstrate that metastatic cancer cells migrate alone [
145]. In humans, the assumption is that cancer cell seeding involves the collective action of a cluster of cancer cells migrating in concert [
146], which is the timepoint at which epithelial-mesenchymal transition (EMT, see
Section 5) comes into play.
Figure 4. Chromosomal instability (CIN) represents a hallmark of cancers that function in the migration and invasion of cancer cells. CIN can be caused by a gain or loss of entire chromosomes, which is referred to as numerical CIN (lilac), or structural reorganization, which is referred to as structural CIN (orange). The loss of heterozygosity (LOH) contributes to numerical and structural CIN, whereby genomic alterations, such as allele loss, impact cancer cell invasiveness. Polyploidy is the existence of additional sets of chromosomes that consequently alter the genetic profile of cancer cells and increase their invasive capacity. Polyploid giant cancer cells (POCC) are seen in numerous cancers and indicate extreme tumorigenic, invasive, and metastatic capacity. Aneuploidy, such as loss of chromosomes (monosomy) or acquired chromosomes (trisomy), can impact cancer cell motility differently. Various gene fusions due to chromosomal rearrangements impact cancer cell invasiveness via different regulatory pathways and mechanisms. Amplification, which is a copy number enhancement of a specific region of the genome, causes elevated gene expression. In this case, when the specific gene is linked to cellular migration, it can increase cancer cell invasion.
Nuclear abnormalities comprise small nuclei (micronuclei) that harbor intact chromosomes or fragments of them. These micronuclei can cause intricate chromosome rearrangements during cancer development and progression [
147]. Nuclear mechanophenotype alteration, such as nuclear envelope blebbing (softening), can induce invaginations that engulf actin and intermediate filaments [
148], which result in epigenetic modifications. In addition, chromosomal abnormalities can lead to polyploidy in cancer cells and, subsequently, to different daughter cancer cells with different mechanophenotypes, which metastasize to varying degrees.
4.2. Exosomes
Exosomes comprise a class of small extracellular vesicles that are liberated by all kinds of cells. They exhibit a size spectrum of 30–200 nm [
149]. Exosomes are formed when the boundary membranes of the endosomes expand towards the lumen and generate multivesicular endosomes. When multivesicular endosomes are trafficked toward the cell surface and merge with the plasma membrane, their intraluminal vesicles are secreted by the cells in the form of exosomes [
150,
151,
152]. Exosomes act in intercellular communication between cancer cells and their microenvironment via the exchange of information through the cargo of the recipient cell, comprising proteins, lipids, DNAs (ssDNA, dsDNA, mtDNA), RNAs (mRNA), and microRNAs (miRNA, long non-coding RNA) [
153]. Exosomes can prevent their cargo, such as miRNAs, from being destroyed by environmental cues, and thus exosome miRNAs can be maintained within human blood plasma and other bodily fluids. Thus, these exosome miRNAs may serve as a non-invasive biomarker for cancer prognosis, treatment, and monitoring, including cervical cancer and malignant glioma [
154,
155]. Exosomes, which are extracellular vesicles holding genetic material, proteins, and lipids, also play a pivotal role in the creation of the pre-metastatic niche [
156]. Increasing stiffening of the TME triggers the release of exosomes from cancer cells via the Akt pathway and Notch pathway activation [
157]. Stiffening of the ECM is associated with activation of Akt, in turn stimulating GTP charging of the small GTPase Rab8, which ultimately propels the release of exosomes [
157]. Exosomes derived from cancer cells exposed to a stiff ECM stimulate tumor growth in an efficient manner. Proteomic profiling revealed that the Notch signaling pathway is induced in cells subjected to exosomes of cancer cells cultured on stiff ECM [
157]. Luminal soluble proteins and membrane proteins of exosomes, such as lipid-anchored proteins, molecules bound to the periphery of the membrane, and transmembrane proteins, can be released into the TME [
158]. The membrane proteins of the exosomes permit them to target specific cells. When they are attached to the targeted cells, exosomes can initiate signaling via receptor-ligand engagement and become endocytosed and/or phagocytosed. Exosomes are able to merge with the membrane of the target cell, thereby liberating their cargo into the cytosol and altering the physiological condition within the cell [
159]. Thereby, the exosomes of a particular cancer cell can impact the performance of surrounding cells, the cellular environment, and the phenotype of remote cells and tissues, which has a strong capacity for systemic implications [
160]. For example, exosomes of cancer cells can impede the immune response against themselves, primarily by causing apoptosis of T lymphocytes [
161]. Exosomes of pancreatic cancer cells target T lymphocytes and interfere with their gene expression profile, e.g., genes such as ATF4, MAPK, and EIF2α, which are involved in the onset of apoptosis, and consequently compromise their antitumor capabilities [
162].
Exosomes can also be captured by adjacent or distant cells, where they are involved in the post-transcriptional regulation of gene expression through the targeting of mRNA. Exosomal miRNAs may fulfill various purposes, such as involvement in inflammatory responses, cell migration, proliferation, apoptosis, autophagy, and epithelial-mesenchymal transition [
163,
164]. Once the exosome miR-132 is captured by endothelial cells, the expression of RabGAP-P120 is decreased through signaling, which encourages the tubularization of endothelial cells [
165].
Exosomes originating from the primary tumor possess a set of integrins on their outer surface that control adhesion of exosomes to specific cell types; this may permit cancer cells to bind the ECM or translocate its cargo into recipient cells to establish organtropism [
166]. New evidence indicates that tumor-derived exosomes can trigger the creation of pre-metastatic niches that facilitate the progression to metastatic disease [
167,
168]. Importantly, exosomal tumor RNAs are found to activate the Toll-like receptor 3 (TLR3) of the alveolar epithelium to activate chemokines (CXCL1, CXCL2, CXCL5, and CXCL12) that are key for neutrophil recruitment and pre-metastatic niche creation inside the lung [
169]. Tumor-derived exosomes may also induce N2 polarization in neutrophils to drive the migration of gastric cancer cells [
170] and neutrophil extracellular traps (NETs) and cause cancer-related thrombosis [
171]. Moreover, exosomes can transport PD-L1 from the primary tumor to other locations in the organism to inhibit the immune defense in the pre-metastatic niche [
172]. In an appropriate pre-metastatic space, cancer cells need to undergo an angiogenic switch to attract various cells that alter the surrounding tissue and create an environment that eases the growth and spread of metastases [
173]. While organ tropism is poorly comprehended, the study of exosomes and pre-metastatic niche creation is advancing knowledge in this field. Targeting exosomes or any other factors that are critical for pre-metastatic niche establishment may be able to avoid the seeding of metastatic cancer, but there is still a long way to go before the formation of pre-metastatic niches is completely clear.
4.3. Cell-Free DNA (cfDNA)
Cell-free (cf) DNA can be released by exosomes that emerge increasingly after a stiffness increase in tumors. As an alternative option to liquid biopsy, circulating cfDNA is steadily secreted from clonal cancer cells into the bloodstream [
174,
175]. Variations in cfDNA can be detected by ultra-deep NGS even at extremely low frequencies (<1%) and have been employed for the early identification of relapses in different tumors (e.g., colorectal cancer, pancreatic cancer, neuroblastoma) and recently also in premalignant lung and bladder forms of the disorder [
176,
177,
178,
179]. Currently, several platform technologies offer adequate sensitivity in detecting circulating tumor (ct) DNA to accurately diagnose lung tumor patients who recur within one year of subclone detection and for accurate screening of premalignant cervical cancer [
176,
180]. However, methods for identifying and profiling ctDNA need to be enhanced, and methods should have improved sensitivity and more specificity with quantitative thresholds to prevent overdiagnosis. An important biological constraint is the quantity of ctDNA collected from early-stage cancer patients, such as when there is even less than 0.1% of ctDNA identified in plasma using digital droplet PCR or NGS techniques [
181]. The enrichment process has to be conducted on the basis of biological or physical characteristics. The results are distorted by cancer-related mutations, which are not limited to cancer patients, and the presence of clonal changes in blood cells resulting from aging and clonal hematopoiesis, which are both considered critical issues [
182,
183]. Genomic driver alterations of cancers in the BRAF, CDKN2A, EGFR, FGFR3, HER2, NF1/2, PIK3CA, RAS, and TP53 genes may also be detected in non-tumor probes. In addition, identifying the tissue of neoplastic lesion origin can be very difficult [
178]. Therefore, the restrictions on the clinical benefit of ctDNA analysis are obvious. Apart from cancer diagnosis, cfDNA can protect cancer cells from being identified and, hence, destroyed by the immune system.
This entry is adapted from the peer-reviewed paper 10.3390/biom14020184