 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | RYUTARO KAJIHARA | -- | 3230 | 2024-02-05 10:44:32 | | | |
| 2 | Peter Tang | Meta information modification | 3230 | 2024-02-06 03:12:04 | | |
Video Upload Options
Both the brain-derived neurotrophic factor (BDNF) and glucocorticoids (GCs) play multiple roles in various aspects of neurons, including cell survival and synaptic function. BDNF and its receptor TrkB are extensively expressed in neurons of the central nervous system (CNS), and the contribution of the BDNF/TrkB system to neuronal function is evident; thus, its downregulation has been considered to be involved in the pathogenesis of Alzheimer’s disease (AD). GCs, stress-related molecules, and glucocorticoid receptors (GRs) are also considered to be associated with AD in addition to mental disorders such as depression. Importantly, a growing body of evidence suggests a close relationship between BDNF/TrkB-mediated signaling and the GCs/GR system in the CNS.
1. Introduction
2. The Role of BDNF in AD
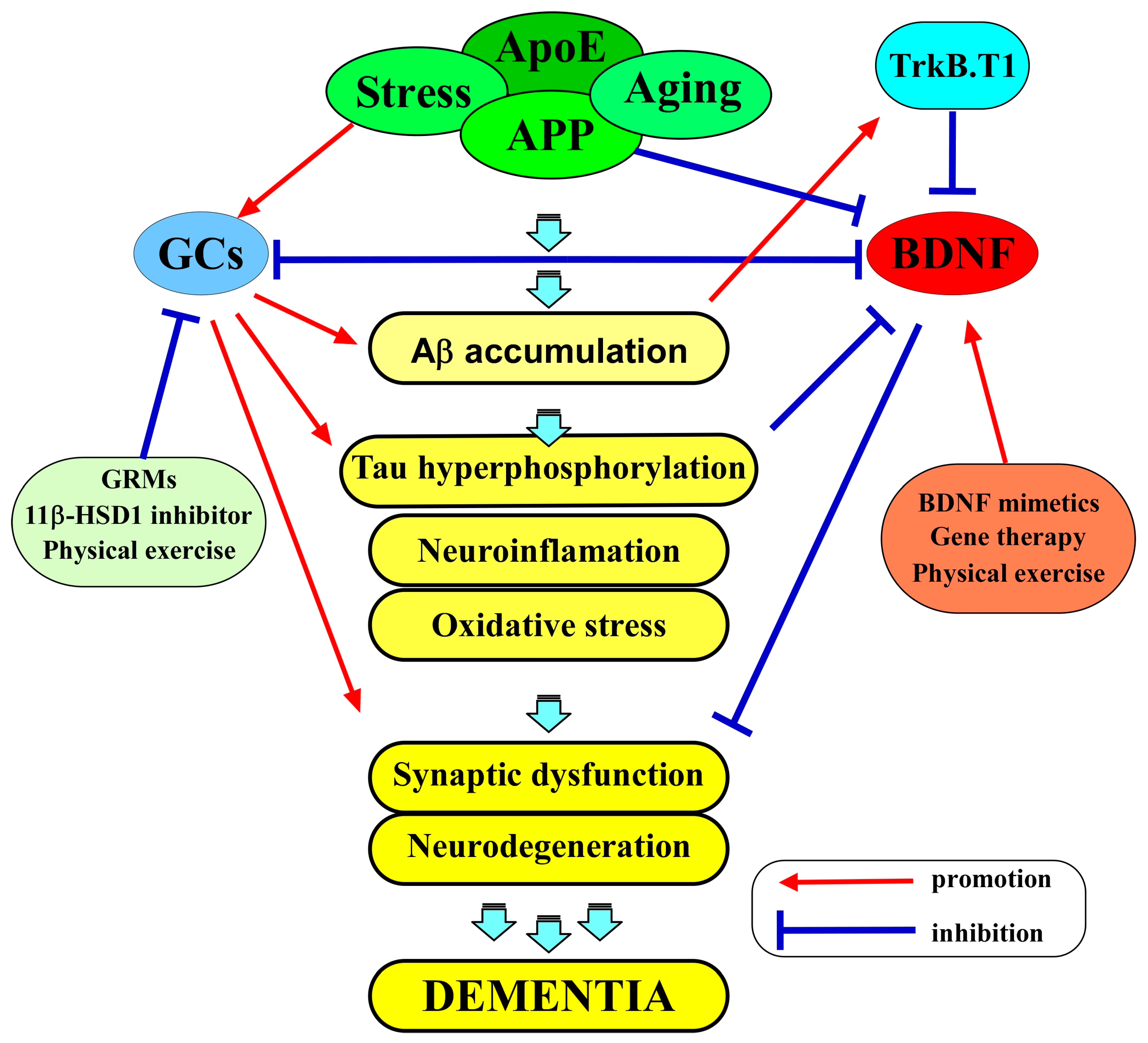
3. The Role of Glucocorticoids in AD
4. BDNF and GCs as Therapeutic Targets in AD
References
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725.
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463.
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806.
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593.
- Pisani, A.; Paciello, F.; Del Vecchio, V.; Malesci, R.; De Corso, E.; Cantone, E.; Fetoni, A.R. The Role of BDNF as a Biomarker in Cognitive and Sensory Neurodegeneration. J. Pers. Med. 2023, 13, 652.
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957.
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front. Mol. Neurosci. 2022, 15, 1028223.
- Kim, E.J.; Pellman, B.; Kim, J.J. Stress effects on the hippocampus: A critical review. Learn. Mem. 2015, 22, 411–416.
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front. Mol. Neurosci. 2023, 16, 1247422.
- Bassil, K.; Krontira, A.C.; Leroy, T.; Escoto, A.I.H.; Snijders, C.; Pernia, C.D.; Pasterkamp, R.J.; de Nijs, L.; van den Hove, D.; Kenis, G.; et al. In vitro modeling of the neurobiological effects of glucocorticoids: A review. Neurobiol. Stress 2023, 23, 100530.
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain 2023, 146, 3969–3990.
- Numakawa, T.; Kajihara, R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life 2023, 13, 647.
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300.
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J. Neurochem. 2002, 82, 1058–1064.
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257.
- Angelucci, F.; Veverova, K.; Katonová, A.; Vyhnalek, M.; Hort, J. Serum PAI-1/BDNF Ratio Is Increased in Alzheimer’s Disease and Correlates with Disease Severity. ACS Omega 2023, 8, 36025–36031.
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a Potential Biomarker of Alzheimer’s Disease: Verification through Assessment of Serum, Cerebrospinal Fluid, and Medial Temporal Lobe Atrophy. Front. Neurol. 2021, 12, 653267.
- Lim, Y.Y.; Laws, S.M.; Perin, S.; Pietrzak, R.H.; Fowler, C.; Masters, C.L.; Maruff, P. BDNF VAL66MET polymorphism and memory decline across the spectrum of Alzheimer’s disease. Genes Brain Behav. 2021, 20, e12724.
- Bessi, V.; Mazzeo, S.; Bagnoli, S.; Padiglioni, S.; Carraro, M.; Piaceri, I.; Bracco, L.; Sorbi, S.; Nacmias, B. The implication of BDNF Val66Met polymorphism in progression from subjective cognitive decline to mild cognitive impairment and Alzheimer’s disease: A 9-year follow-up study. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 471–482.
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269.
- del Toro, D.; Canals, J.M.; Ginés, S.; Kojima, M.; Egea, G.; Alberch, J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 2006, 26, 12748–12757.
- Wang, Y.; Li, O.; Li, N.; Sha, Z.; Zhao, Z.; Xu, J. Association between the BDNF Val66Met polymorphism and major depressive disorder: A systematic review and meta-analysis. Front. Psychiatry 2023, 14, 1143833.
- Brown, D.T.; Vickers, J.C.; Stuart, K.E.; Cechova, K.; Ward, D.D. The BDNF Val66Met Polymorphism Modulates Resilience of Neurological Functioning to Brain Ageing and Dementia: A Narrative Review. Brain Sci. 2020, 10, 195.
- Eggert, S.; Kins, S.; Endres, K.; Brigadski, T. Brothers in arms: ProBDNF/BDNF and sAPPα/Aβ-signaling and their common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in the progression of Alzheimer’s disease. Biol. Chem. 2022, 403, 43–71.
- Zheng, Z.; Sabirzhanov, B.; Keifer, J. Oligomeric amyloid- inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J. Biol. Chem. 2010, 285, 34708–34717.
- Yan, P.; Xue, Z.; Li, D.; Ni, S.; Wang, C.; Jin, X.; Zhou, D.; Li, X.; Zhao, X.; Chen, X.; et al. Dysregulated CRTC1-BDNF signaling pathway in the hippocampus contributes to Aβ oligomer-induced long-term synaptic plasticity and memory impairment. Exp. Neurol. 2021, 345, 113812.
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer’s disease induced by aβ1-42. PLoS ONE 2015, 10, e0122415.
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci. Ther. 2019, 25, 303–313.
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 991–1001.
- Jerónimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lérias, S.; Caetano, A.P.; Buée-Scherrer, V.; Castrén, E.; Valente, C.A.; Blum, D.; Sebastião, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18.
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731.
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1β administrations differentially modulate neuroimmune and neurotrophic systems: Possible implications for neuroprotection and neurodegeneration. J. Neuroinflamm. 2013, 10, 59.
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol. Aging 2008, 29, 1380–1393.
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharmacol. Res. 2020, 158, 104865.
- Oreshko, A.S.; Rodnyy, A.Y.; Bazovkina, D.V.; Naumenko, V.S. Effects of central administration of the human Tau protein on the Bdnf, Trkb, p75, Mapt, Bax and Bcl-2 genes expression in the mouse brain. Vavilovskii Zhurnal Genet. Selektsii 2023, 27, 342–348.
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518.
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221.
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551.
- Laczó, J.; Cechova, K.; Parizkova, M.; Lerch, O.; Andel, R.; Matoska, V.; Kaplan, V.; Matuskova, V.; Nedelska, Z.; Vyhnalek, M.; et al. The Combined Effect of APOE and BDNF Val66Met Polymorphisms on Spatial Navigation in Older Adults. J. Alzheimers Dis. 2020, 78, 1473–1492.
- Pietzuch, M.; Bindoff, A.; Jamadar, S.; Vickers, J.C. Interactive effects of the APOE and BDNF polymorphisms on functional brain connectivity: The Tasmanian Healthy Brain Project. Sci. Rep. 2021, 11, 14514.
- Viho, E.M.G.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; van Weert, L.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266–276.
- Koning, A.; Buurstede, J.C.; van Weert, L.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J. Endocr. Soc. 2019, 3, 1917–1930.
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104568.
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37.
- Dioli, C.; Papadimitriou, G.; Megalokonomou, A.; Marques, C.; Sousa, N.; Sotiropoulos, I. Chronic Stress, Depression, and Alzheimer’s Disease: The Triangle of Oblivion. Adv. Exp. Med. Biol. 2023, 1423, 303–315.
- Forget, H.; Lacroix, A.; Somma, M.; Cohen, H. Cognitive decline in patients with Cushing’s syndrome. J. Int. Neuropsychol. Soc. 2000, 6, 20–29.
- Notarianni, E. Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer’s disease initiation and development. Curr. Alzheimer Res. 2013, 10, 714–731.
- Zheng, B.; Tal, R.; Yang, Z.; Middleton, L.; Udeh-Momoh, C. Cortisol hypersecretion and the risk of Alzheimer’s disease: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101171.
- Klyubin, I.; Ondrejcak, T.; Hu, N.-W.; Rowan, M.J. Glucocorticoids, synaptic plasticity and Alzheimer’s disease. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100365.
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378–4394.
- Kulstad, J.J.; McMillan, P.J.; Leverenz, J.B.; Cook, D.G.; Green, P.S.; Peskind, E.R.; Wilkinson, C.W.; Farris, W.; Mehta, P.D.; Craft, S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J. Neuropathol. Exp. Neurol. 2005, 64, 139–146.
- Ding, S.; Yang, L.; Huang, L.; Kong, L.; Chen, M.; Su, Y.; Li, X.; Dong, X.; Han, Y.; Li, W.; et al. Chronic glucocorticoid exposure accelerates Aβ generation and neurotoxicity by activating calcium-mediated CN-NFAT1 signaling in hippocampal neurons in APP/PS1 mice. Food Chem. Toxicol. 2022, 168, 113407.
- Siegel, G.; Gerber, H.; Koch, P.; Bruestle, O.; Fraering, P.C.; Rajendran, L. The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides. Cell Rep. 2017, 19, 1967–1976.
- Wang, Y.; Li, M.; Tang, J.; Song, M.; Xu, X.; Xiong, J.; Li, J.; Bai, Y. Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 2011, 152, 2704–2715.
- Sotiropoulos, I.; Catania, C.; Riedemann, T.; Fry, J.P.; Breen, K.C.; Michaelidis, T.M.; Almeida, O.F. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J. Neurochem. 2008, 107, 385–397.
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847.
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83.
- Yi, J.H.; Brown, C.; Whitehead, G.; Piers, T.; Lee, Y.S.; Perez, C.M.; Regan, P.; Whitcomb, D.J.; Cho, K. Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol. Res. 2017, 121, 42–51.
- Milligan Armstrong, A.; Porter, T.; Quek, H.; White, A.; Haynes, J.; Jackaman, C.; Villemagne, V.; Munyard, K.; Laws, S.M.; Verdile, G.; et al. Chronic stress and Alzheimer’s disease: The interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2209–2228.
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2990.
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347.
- Harris-White, M.E.; Chu, T.; Miller, S.A.; Simmons, M.; Teter, B.; Nash, D.; Cole, G.M.; Frautschy, S.A. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and A beta clearance in vitro and in vivo. Neurochem. Int. 2001, 39, 435–448.
- Sharma, V.K.; Singh, T.G. Navigating Alzheimer’s Disease via Chronic Stress: The Role of Glucocorticoids. Curr. Drug Targets 2020, 21, 433–444.
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232.
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101.
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The Potential Role of the NLRP3 Inflammasome Activation as a Link between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front. Cell. Neurosci. 2019, 13, 73.
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954.
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 2017, 12, 7–12.
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583.
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting both BDNF/TrkB pathway and delta-secretase for treating Alzheimer’s disease. Neuropharmacology 2021, 197, 108737.
- Pak, M.E.; Yang, H.J.; Li, W.; Kim, J.K.; Go, Y. Yuk-Gunja-Tang attenuates neuronal death and memory impairment via ERK/CREB/BDNF signaling in the hippocampi of experimental Alzheimer’s disease model. Front. Pharmacol. 2022, 13, 1014840.
- Gupta, V.; Chitranshi, N.; Gupta, V.; You, Y.; Rajput, R.; Paulo, J.A.; Mirzaei, M.; van den Buuse, M.; Graham, S.L. TrkB Receptor Agonist 7,8 Dihydroxyflavone is Protective Against the Inner Retinal Deficits Induced by Experimental Glaucoma. Neuroscience 2022, 490, 36–48.
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer’s disease progression. Front. Mol. Neurosci. 2023, 16, 1150399.
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602.
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907.
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296.
- Kim, D.; Cho, J.; Kang, H. Protective effect of exercise training against the progression of Alzheimer’s disease in 3xTg-AD mice. Behav. Brain Res. 2019, 374, 112105.
- Xu, L.; Zhu, L.; Zhu, L.; Chen, D.; Cai, K.; Liu, Z.; Chen, A. Moderate Exercise Combined with Enriched Environment Enhances Learning and Memory through BDNF/TrkB Signaling Pathway in Rats. Int. J. Environ. Res. Public Health 2021, 18, 8283.
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692.
- Zhang, J.; Zheng, X.; Zhao, Z. A systematic review and meta-analysis on the efficacy outcomes of selective serotonin reuptake inhibitors in depression in Alzheimer’s disease. BMC Neurol. 2023, 23, 210.
- Casarotto, P.; Umemori, J.; Castrén, E. BDNF receptor TrkB as the mediator of the antidepressant drug action. Front. Mol. Neurosci. 2022, 15, 1032224.
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e1219.
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181.
- Håkansson, K.; Ledreux, A.; Daffner, K.; Terjestam, Y.; Bergman, P.; Carlsson, R.; Kivipelto, M.; Winblad, B.; Granholm, A.C.; Mohammed, A.K. BDNF Responses in Healthy Older Persons to 35 Minutes of Physical Exercise, Cognitive Training, and Mindfulness: Associations with Working Memory Function. J. Alzheimers Dis. 2017, 55, 645–657.
- Angelucci, F.; Peppe, A.; Carlesimo, G.A.; Serafini, F.; Zabberoni, S.; Barban, F.; Shofany, J.; Caltagirone, C.; Costa, A. A pilot study on the effect of cognitive training on BDNF serum levels in individuals with Parkinson’s disease. Front. Hum. Neurosci. 2015, 9, 130.
- Gomutbutra, P.; Srikamjak, T.; Sapinun, L.; Kunaphanh, S.; Yingchankul, N.; Apaijai, N.; Shinlapawittayatorn, K.; Phuackchantuck, R.; Chattipakorn, N.; Chattipakorn, S. Effect of intensive weekend mindfulness-based intervention on BDNF, mitochondria function, and anxiety. A randomized, crossover clinical trial. Compr. Psychoneuroendocrinol. 2022, 11, 100137.
- Canet, G.; Hernandez, C.; Zussy, C.; Chevallier, N.; Desrumaux, C.; Givalois, L. Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets. Front. Aging Neurosci. 2019, 11, 269.
- Watermeyer, T.; Robb, C.; Gregory, S.; Udeh-Momoh, C. Therapeutic implications of hypothalamic-pituitaryadrenal-axis modulation in Alzheimer’s disease: A narrative review of pharmacological and lifestyle interventions. Front. Neuroendocrinol. 2021, 60, 100877.
- Sooy, K.; Webster, S.P.; Noble, J.; Binnie, M.; Walker, B.R.; Seckl, J.R.; Yau, J.L. Partial deficiency or short-term inhibition of 11beta-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J. Neurosci. 2010, 30, 13867–13872.
- Sooy, K.; Noble, J.; McBride, A.; Binnie, M.; Yau, J.L.; Seckl, J.R.; Walker, B.R.; Webster, S.P. Cognitive and Disease-Modifying Effects of 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibition in Male Tg2576 Mice, a Model of Alzheimer’s Disease. Endocrinology 2015, 156, 4592–4603.
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 2013, 74, 357–366.
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol. Aging 2016, 45, 109–122.
- da Costa Daniele, T.M.; de Bruin, P.F.C.; de Matos, R.S.; de Bruin, G.S.; Maia Chaves, C.J.; de Bruin, V.M.S. Exercise effects on brain and behavior in healthy mice, Alzheimer’s disease and Parkinson’s disease model-A systematic review and meta-analysis. Behav. Brain Res. 2020, 383, 112488.
- Bashiri, H.; Enayati, M.; Bashiri, A.; Salari, A.A. Swimming exercise improves cognitive and behavioral disorders in male NMRI mice with sporadic Alzheimer-like disease. Physiol. Behav. 2020, 223, 113003.
- Campos, H.C.; Ribeiro, D.E.; Hashiguchi, D.; Glaser, T.; Milanis, M.D.S.; Gimenes, C.; Suchecki, D.; Arida, R.M.; Ulrich, H.; Monteiro Longo, B. Neuroprotective effects of resistance physical exercise on the APP/PS1 mouse model of Alzheimer’s disease. Front. Neurosci. 2023, 17, 1132825.
- Irazoki, E.; Contreras-Somoza, L.M.; Toribio-Guzmán, J.M.; Jenaro-Río, C.; van der Roest, H.; Franco-Martín, M.A. Technologies for Cognitive Training and Cognitive Rehabilitation for People With Mild Cognitive Impairment and Dementia. A Systematic Review. Front. Psychol. 2020, 11, 648.
- Cutuli, D.; Decandia, D.; Giacovazzo, G.; Coccurello, R. Physical Exercise as Disease-Modifying Alternative against Alzheimer’s Disease: A Gut-Muscle-Brain Partnership. Int. J. Mol. Sci. 2023, 24, 4686.

