Both the brain-derived neurotrophic factor (BDNF) and glucocorticoids (GCs) play multiple roles in various aspects of neurons, including cell survival and synaptic function. BDNF and its receptor TrkB are extensively expressed in neurons of the central nervous system (CNS), and the contribution of the BDNF/TrkB system to neuronal function is evident; thus, its downregulation has been considered to be involved in the pathogenesis of Alzheimer’s disease (AD). GCs, stress-related molecules, and glucocorticoid receptors (GRs) are also considered to be associated with AD in addition to mental disorders such as depression. Importantly, a growing body of evidence suggests a close relationship between BDNF/TrkB-mediated signaling and the GCs/GR system in the CNS.
- brain-derived neurotrophic factor
- TrkB
- intracellular signaling
- synaptic plasticity
- glucocorticoids
- GR
- depression
- Alzheimer’s disease
1. Introduction
2. The Role of BDNF in AD
AD is a multifactorial neurodegenerative disorder characterized by progressive cognitive decline, synaptic dysfunction, and memory impairment. Although the exact cause of AD is not fully understood, the amyloid hypothesis is widely recognized as a theory that provides a framework for understanding the pathogenesis of the disease [63][11]. It proposes that the accumulation of beta-amyloid (Aβ) plaques in the brain is a central event in the development of the disease. Amyloid precursor protein (APP) is a transmembrane protein that is present in many cells, including neurons. In the amyloidogenic pathway, APP is first cleaved by β-secretase (BACE-1), and then the remaining fragment is cleaved by γ-secretase, releasing Aβ peptides of various lengths, including the longer Aβ42 form, which is particularly prone to aggregation. The aggregated Aβ is thought to have neurotoxic effects, leading to abnormal phosphorylation of the tau protein, the subsequent formation of neurofibrillary tangles, the death of neurons, and the progressive cognitive decline observed in AD (Figure 1) [63][11]. While the disease’s hallmark pathological features are the accumulation of Aβ plaques and neurofibrillary tangles, increasing attention has been directed toward understanding the role of BDNF dysregulation in AD pathophysiology [9,64][9][12].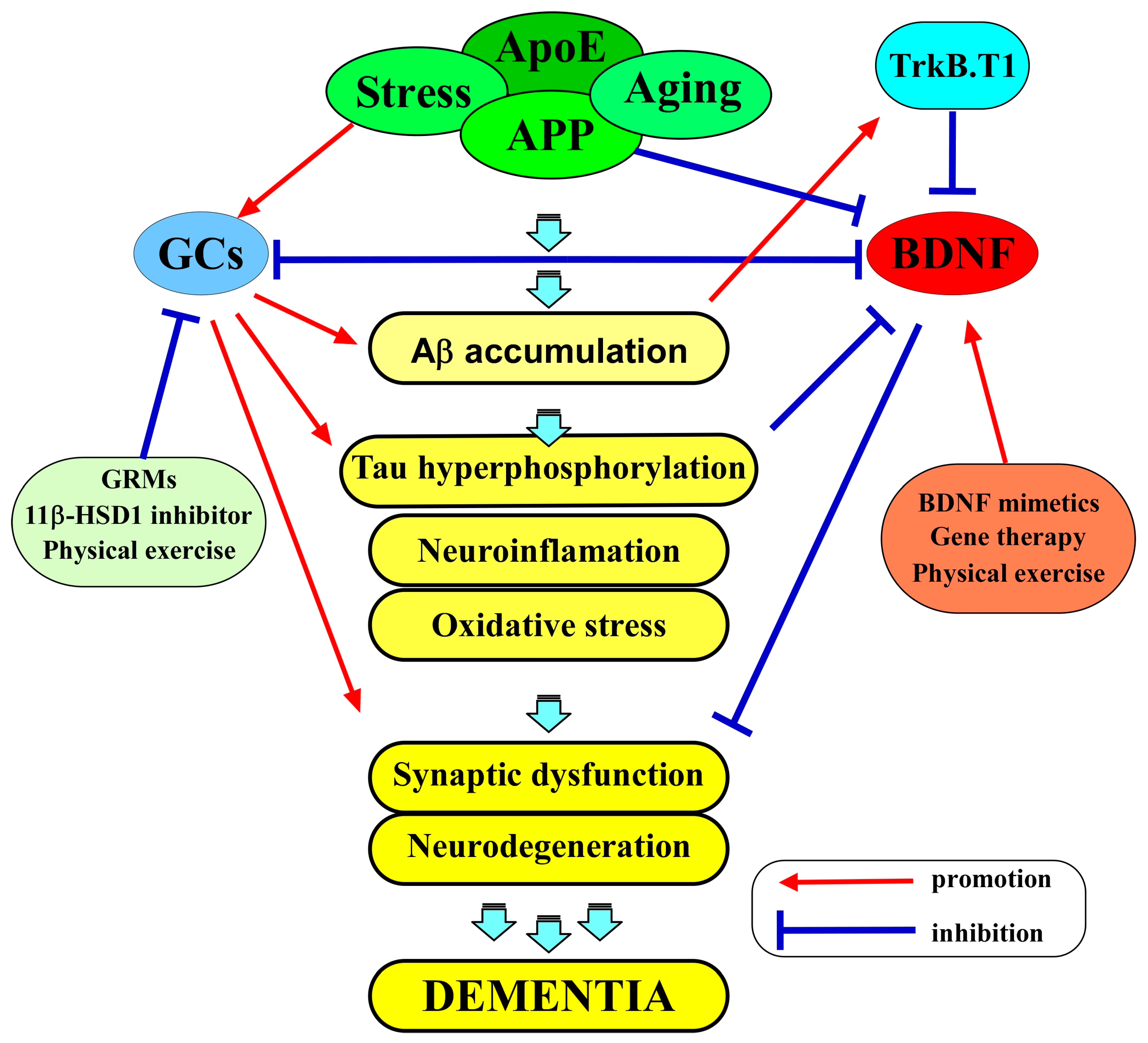
3. The Role of Glucocorticoids in AD
The role of glucocorticoids in the pathophysiology of AD is also a subject of growing interest and significance. The central concept for understanding the molecular relationship between glucocorticoids and AD is the presence of GRs in the brain. These receptors, primarily located in the hippocampus and prefrontal cortex, are known to mediate the effects of GCs on various physiological processes, including metabolism, immune response, and neural plasticity [93,94][43][44]. Recent research has demonstrated that the expression of GRs in key brain regions is linked to vulnerability to AD pathology [95][45]. The interaction between GCs and these receptors is complex. Under normal physiological conditions, GCs play a crucial role in synaptic plasticity, cognitive function, and memory consolidation [96][46]. However, chronic exposure to elevated GC levels, a common consequence of chronic stress, can lead to GR dysregulation [97][47]. This dysregulation may result in a hyperactive stress response and potentially contribute to AD pathogenesis [97][47]. Epidemiological and clinical investigations have supplied evidence that supports the correlation between chronic exposure to GCs and an elevated risk of developing AD. For example, prolonged exposure to GCs, observed in clinical conditions such as Cushing’s syndrome or extended use of corticosteroid medications, has been linked to cognitive impairment resembling the characteristics of AD [98,99][48][49]. Moreover, Zheng et al. (2020) recently demonstrated that AD patients exhibited higher morning cortisol levels compared to controls, and elevated cortisol levels were correlated with accelerated cognitive decline in individuals with mild cognitive impairment (MCI) [100][50]. Aβ plaques and hyperphosphorylated tau tangles are the hallmarks of AD pathology. Studies have unveiled intriguing links between GCs and the accumulation of these toxic proteins [101,102][51][52]. Notably, GCs appear to influence the levels of both Aβ and tau through distinct mechanisms. Research indicates that GCs can promote the production of Aβ peptides, particularly the more aggregation-prone Aβ42 isoform [103][53]. This effect may be mediated through the modulation of enzymes involved in Aβ production, such as β-secretase [104,105][54][55]. Furthermore, GCs have been implicated in impairing the clearance of Aβ from the brain, potentially by interfering with several Aβ-degrading proteases, such as insulin-degrading enzymes and matrix metalloproteinase-9 [106][56]. In the context of tau pathology, GCs have been associated with the hyperphosphorylation of tau protein [102][52]. Chronic GC exposure is known to activate kinases responsible for tau phosphorylation, including GSK3, CDK5, and ERK1/2, leading to the formation of neurofibrillary tangles [107,108,109,110][57][58][59][60]. This tau pathology is closely tied to synaptic dysfunction and neurodegeneration in AD [102][52]. Neuroinflammation is another crucial element in understanding the role of GCs in AD. Microglia, the resident immune cells of the CNS, play a pivotal role in maintaining brain homeostasis and responding to pathological insults. Recent research has revealed a multifaceted connection between GCs and microglia activation [95,111][45][61]. Chronic exposure to elevated GC levels, as seen in conditions of chronic stress, may result in an overactivation of microglia [112][62]. This hyperactivity can lead to a pro-inflammatory state, characterized by the release of pro-inflammatory cytokines, reactive oxygen species (ROS), and other neurotoxic molecules [112][62]. Such neuroinflammatory responses are detrimental to neuronal health and have been closely associated with the progression of AD [112][62]. Additionally, GCs can modulate the microglial phenotype. Under normal conditions, microglia can exhibit both pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes, depending on the context [113][63]. Dysregulated GC signaling has been shown to skew microglia toward the pro-inflammatory state, exacerbating neuroinflammation in AD [113][63]. Moreover, microglia-mediated degradation of Aβ, which is crucial for Aβ clearance, may be impaired under conditions of GC dysregulation [114][64]. Oxidative stress is another prominent feature of neuroinflammation and the pathophysiology of AD. GCs have been implicated in modulating oxidative stress pathways, further linking them to neuroinflammatory processes [115][65]. Elevated GC levels can promote the generation of ROS and reduce the brain’s antioxidant defenses [116][66]. This imbalance can lead to oxidative damage to cells, proteins, and lipids [117][67]. Oxidative stress not only directly contributes to neurodegeneration but also exacerbates neuroinflammation by activating microglia and promoting the release of pro-inflammatory mediators [118][68]. Furthermore, the oxidative damage inflicted by GC-induced oxidative stress may also play a role in the formation of Aβ plaques and tau tangles [119][69]. Oxidatively modified proteins are more prone to aggregation, and they may contribute to the seeding and propagation of Aβ and tau pathology [119][69]. Understanding these cellular mechanisms is vital to appreciating the intricate relationship between GC dysregulation and neuroinflammation in AD. Dysregulated GC signaling can create a microenvironment that favors neuroinflammation and oxidative stress, both of which are detrimental to neuronal health. However, it is essential to recognize that while GCs are a piece of this puzzle, they do not act in isolation. Genetic factors, other environmental stressors, and the interplay between different pathological processes in AD must also be considered in the complex pathophysiology of this disease.4. BDNF and GCs as Therapeutic Targets in AD
Given the pivotal role of BDNF in neuronal survival, synaptic plasticity, and cognitive function, BDNF dysregulation presents an attractive target for therapeutic interventions in AD. Strategies aimed at restoring BDNF levels and function, either by promoting its production or enhancing its signaling, hold promise for mitigating the cognitive deficits associated with AD. Several experimental approaches, such as BDNF mimetics [120[70][71][72][73][74][75],121,122,123,124,125], gene therapy [126[76][77],127], and lifestyle interventions like physical activity [128[78][79][80],129,130], are being explored as potential ways to address BDNF dysregulation and its consequences (Figure 1). BDNF mimetics are compounds that mimic the actions of BDNF or enhance its receptor binding, promoting neuroprotection and synaptic plasticity. One of these BDNF-enhancing compounds is 7,8-dihydroxyflavone (7,8-DHF), a selective TrkB agonist, as demonstrated by Jang et al. (2010) [131][81]. Additionally, Yuk-Gunja-Tang (YG), a Korean traditional medicine, has the capacity to enhance the endogenous expression of BDNF [123][73]. Numerous preclinical studies have provided evidence of the effectiveness of these agents in animal models of AD [120,121,122,123,124,125][70][71][72][73][74][75]. Importantly, patients with AD are often prescribed antidepressants, including selective serotonin reuptake inhibitors (SSRIs), to alleviate the depressive symptoms of AD [132][82]. At the same time, it is well known that SSRIs exert their effects by acting on monoamine transporters, which subsequently results in the activation of BDNF/TrkB signaling [133][83]. Interestingly, Casarotto et al. (2021) recently identified that SSRIs also directly bind to TrkB receptors, inducing an allosteric potentiation of TrkB signaling [134][84]. These findings provide support for the notion that BDNF/TrkB signaling serves as the direct target for antidepressant drugs, playing a role in mediating their therapeutic effects in AD patients. In addition to the pharmacological approaches, lifestyle interventions such as physical exercise and cognitive stimulation have been shown to boost BDNF levels in the brain [130][80]. A meta-analysis of randomized controlled trials, as reported by Jia et al. (2019), indicated that exercise interventions were linked to significant enhancements in global cognitive function among patients with mild-to-moderate AD [135][85]. Likewise, research has shown that exercise training can enhance BDNF expression and cognitive function and stimulate neuroplasticity in animal models of AD [128][78]. Another clinical study also demonstrated that healthy older adults exposed to 35-minute sessions of physical exercise, cognitive training, and mindfulness practice increased serum BDNF levels [136][86]. Moreover, patients with Parkinson’s disease undergoing cognitive stimulation displayed increased serum BDNF levels as compared to the placebo group [137][87]. Additionally, Gomutbutra et al. (2022) recently showed that even a brief period of mindfulness-based intervention (MBI) can elevate serum BDNF levels and decrease anxiety in healthy, meditation-naïve females in a randomized, crossover clinical trial [138][88]. Recognizing the influence of GC dysregulation in AD pathophysiology opens up possibilities for novel therapeutic interventions. Strategies that aim to modulate GC activity represent a promising approach (Figure 1). These include pharmacological interventions to normalize cortisol levels or reduce the sensitivity of glucocorticoid receptors. 11β-HSD1 is a pivotal enzyme that is responsible for the intracellular conversion of inactive cortisone into its active form, cortisol, in humans (or 11-dehydrocortisone into corticosterone in rodents). Consequently, inhibiting 11β-HSD1 results in a decrease in cortisol levels in humans (or CORT levels in rodents) [139,140][89][90]. Sooy et al. (2010) showed that UE1961, an inhibitor of 11β-HSD1, demonstrated a significant enhancement in spatial memory performance in aged mice [141][91]. Moreover, the researchers showed that the administration of another inhibitor, UE2316, led to a decrease in Aβ plaques within the cortex of aged Tg2576 mice [142][92]. This reduction was concurrent with an elevation in insulin-degrading enzyme (one of the Aβ-degrading proteases) levels, which, in turn, resulted in memory improvements [142][92]. In addition to 11β-HSD1 inhibitors, selective GR modulators (GRMs) are designed to specifically inhibit GR activity in AD. A study demonstrated that treatment with CORT108297, one of the GRMs, led to a reduction in the levels of APP C-terminal fragments in the 3xTg-AD mouse mode; [143][93]. Another study also showed that mice that were administered CORT108297 via intraperitoneal injection exhibited a complete reversal of memory deficits, as evaluated through the T-maze test [144][94]. Besides pharmacological approaches, non-pharmacological interventions aimed at fostering resilience to stress and improving cognitive function could potentially offer benefits in the management of AD [145][95]. Stress management techniques, such as mindfulness-based interventions and cognitive-behavioral therapy, may be explored as preventive measures for individuals at risk of AD [140][90]. These approaches could reduce stress-related GR fluctuations and potentially delay disease onset [146,147][96][97]. Lifestyle modifications, including regular physical exercise, a balanced diet, and adequate sleep, may also have a significant impact on GC regulation [148][98]. These factors can help maintain a healthy stress response system and mitigate the effects of chronic stress on neuroinflammation, oxidative stress, and Aβ/Tau pathology [147,149][97][99]. BDNF and GCs stand as pivotal factors in the pathophysiology of AD. Their roles in cognitive function, synaptic plasticity, and Aβ/Tau pathology underscore their significance in understanding the disease and their potential as therapeutic targets. Investigating the intricate relationship between BDNF and GCs in AD remains an active area of research, offering hope for novel interventions aimed at mitigating cognitive decline and improving the lives of individuals affected by this challenging neurodegenerative disorder.References
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725.
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463.
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806.
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593.
- Pisani, A.; Paciello, F.; Del Vecchio, V.; Malesci, R.; De Corso, E.; Cantone, E.; Fetoni, A.R. The Role of BDNF as a Biomarker in Cognitive and Sensory Neurodegeneration. J. Pers. Med. 2023, 13, 652.
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957.
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front. Mol. Neurosci. 2022, 15, 1028223.
- Kim, E.J.; Pellman, B.; Kim, J.J. Stress effects on the hippocampus: A critical review. Learn. Mem. 2015, 22, 411–416.
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front. Mol. Neurosci. 2023, 16, 1247422.
- Bassil, K.; Krontira, A.C.; Leroy, T.; Escoto, A.I.H.; Snijders, C.; Pernia, C.D.; Pasterkamp, R.J.; de Nijs, L.; van den Hove, D.; Kenis, G.; et al. In vitro modeling of the neurobiological effects of glucocorticoids: A review. Neurobiol. Stress 2023, 23, 100530.
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain 2023, 146, 3969–3990.
- Numakawa, T.; Kajihara, R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life 2023, 13, 647.
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300.
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J. Neurochem. 2002, 82, 1058–1064.
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257.
- Angelucci, F.; Veverova, K.; Katonová, A.; Vyhnalek, M.; Hort, J. Serum PAI-1/BDNF Ratio Is Increased in Alzheimer’s Disease and Correlates with Disease Severity. ACS Omega 2023, 8, 36025–36031.
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a Potential Biomarker of Alzheimer’s Disease: Verification through Assessment of Serum, Cerebrospinal Fluid, and Medial Temporal Lobe Atrophy. Front. Neurol. 2021, 12, 653267.
- Lim, Y.Y.; Laws, S.M.; Perin, S.; Pietrzak, R.H.; Fowler, C.; Masters, C.L.; Maruff, P. BDNF VAL66MET polymorphism and memory decline across the spectrum of Alzheimer’s disease. Genes Brain Behav. 2021, 20, e12724.
- Bessi, V.; Mazzeo, S.; Bagnoli, S.; Padiglioni, S.; Carraro, M.; Piaceri, I.; Bracco, L.; Sorbi, S.; Nacmias, B. The implication of BDNF Val66Met polymorphism in progression from subjective cognitive decline to mild cognitive impairment and Alzheimer’s disease: A 9-year follow-up study. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 471–482.
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269.
- del Toro, D.; Canals, J.M.; Ginés, S.; Kojima, M.; Egea, G.; Alberch, J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 2006, 26, 12748–12757.
- Wang, Y.; Li, O.; Li, N.; Sha, Z.; Zhao, Z.; Xu, J. Association between the BDNF Val66Met polymorphism and major depressive disorder: A systematic review and meta-analysis. Front. Psychiatry 2023, 14, 1143833.
- Brown, D.T.; Vickers, J.C.; Stuart, K.E.; Cechova, K.; Ward, D.D. The BDNF Val66Met Polymorphism Modulates Resilience of Neurological Functioning to Brain Ageing and Dementia: A Narrative Review. Brain Sci. 2020, 10, 195.
- Eggert, S.; Kins, S.; Endres, K.; Brigadski, T. Brothers in arms: ProBDNF/BDNF and sAPPα/Aβ-signaling and their common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in the progression of Alzheimer’s disease. Biol. Chem. 2022, 403, 43–71.
- Zheng, Z.; Sabirzhanov, B.; Keifer, J. Oligomeric amyloid- inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J. Biol. Chem. 2010, 285, 34708–34717.
- Yan, P.; Xue, Z.; Li, D.; Ni, S.; Wang, C.; Jin, X.; Zhou, D.; Li, X.; Zhao, X.; Chen, X.; et al. Dysregulated CRTC1-BDNF signaling pathway in the hippocampus contributes to Aβ oligomer-induced long-term synaptic plasticity and memory impairment. Exp. Neurol. 2021, 345, 113812.
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer’s disease induced by aβ1-42. PLoS ONE 2015, 10, e0122415.
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci. Ther. 2019, 25, 303–313.
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 991–1001.
- Jerónimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lérias, S.; Caetano, A.P.; Buée-Scherrer, V.; Castrén, E.; Valente, C.A.; Blum, D.; Sebastião, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18.
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731.
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1β administrations differentially modulate neuroimmune and neurotrophic systems: Possible implications for neuroprotection and neurodegeneration. J. Neuroinflamm. 2013, 10, 59.
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol. Aging 2008, 29, 1380–1393.
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharmacol. Res. 2020, 158, 104865.
- Oreshko, A.S.; Rodnyy, A.Y.; Bazovkina, D.V.; Naumenko, V.S. Effects of central administration of the human Tau protein on the Bdnf, Trkb, p75, Mapt, Bax and Bcl-2 genes expression in the mouse brain. Vavilovskii Zhurnal Genet. Selektsii 2023, 27, 342–348.
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518.
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221.
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551.
- Laczó, J.; Cechova, K.; Parizkova, M.; Lerch, O.; Andel, R.; Matoska, V.; Kaplan, V.; Matuskova, V.; Nedelska, Z.; Vyhnalek, M.; et al. The Combined Effect of APOE and BDNF Val66Met Polymorphisms on Spatial Navigation in Older Adults. J. Alzheimers Dis. 2020, 78, 1473–1492.
- Pietzuch, M.; Bindoff, A.; Jamadar, S.; Vickers, J.C. Interactive effects of the APOE and BDNF polymorphisms on functional brain connectivity: The Tasmanian Healthy Brain Project. Sci. Rep. 2021, 11, 14514.
- Viho, E.M.G.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; van Weert, L.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266–276.
- Koning, A.; Buurstede, J.C.; van Weert, L.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J. Endocr. Soc. 2019, 3, 1917–1930.
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104568.
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37.
- Dioli, C.; Papadimitriou, G.; Megalokonomou, A.; Marques, C.; Sousa, N.; Sotiropoulos, I. Chronic Stress, Depression, and Alzheimer’s Disease: The Triangle of Oblivion. Adv. Exp. Med. Biol. 2023, 1423, 303–315.
- Forget, H.; Lacroix, A.; Somma, M.; Cohen, H. Cognitive decline in patients with Cushing’s syndrome. J. Int. Neuropsychol. Soc. 2000, 6, 20–29.
- Notarianni, E. Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer’s disease initiation and development. Curr. Alzheimer Res. 2013, 10, 714–731.
- Zheng, B.; Tal, R.; Yang, Z.; Middleton, L.; Udeh-Momoh, C. Cortisol hypersecretion and the risk of Alzheimer’s disease: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101171.
- Klyubin, I.; Ondrejcak, T.; Hu, N.-W.; Rowan, M.J. Glucocorticoids, synaptic plasticity and Alzheimer’s disease. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100365.
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378–4394.
- Kulstad, J.J.; McMillan, P.J.; Leverenz, J.B.; Cook, D.G.; Green, P.S.; Peskind, E.R.; Wilkinson, C.W.; Farris, W.; Mehta, P.D.; Craft, S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J. Neuropathol. Exp. Neurol. 2005, 64, 139–146.
- Ding, S.; Yang, L.; Huang, L.; Kong, L.; Chen, M.; Su, Y.; Li, X.; Dong, X.; Han, Y.; Li, W.; et al. Chronic glucocorticoid exposure accelerates Aβ generation and neurotoxicity by activating calcium-mediated CN-NFAT1 signaling in hippocampal neurons in APP/PS1 mice. Food Chem. Toxicol. 2022, 168, 113407.
- Siegel, G.; Gerber, H.; Koch, P.; Bruestle, O.; Fraering, P.C.; Rajendran, L. The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides. Cell Rep. 2017, 19, 1967–1976.
- Wang, Y.; Li, M.; Tang, J.; Song, M.; Xu, X.; Xiong, J.; Li, J.; Bai, Y. Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 2011, 152, 2704–2715.
- Sotiropoulos, I.; Catania, C.; Riedemann, T.; Fry, J.P.; Breen, K.C.; Michaelidis, T.M.; Almeida, O.F. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J. Neurochem. 2008, 107, 385–397.
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847.
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83.
- Yi, J.H.; Brown, C.; Whitehead, G.; Piers, T.; Lee, Y.S.; Perez, C.M.; Regan, P.; Whitcomb, D.J.; Cho, K. Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol. Res. 2017, 121, 42–51.
- Milligan Armstrong, A.; Porter, T.; Quek, H.; White, A.; Haynes, J.; Jackaman, C.; Villemagne, V.; Munyard, K.; Laws, S.M.; Verdile, G.; et al. Chronic stress and Alzheimer’s disease: The interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2209–2228.
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2990.
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347.
- Harris-White, M.E.; Chu, T.; Miller, S.A.; Simmons, M.; Teter, B.; Nash, D.; Cole, G.M.; Frautschy, S.A. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and A beta clearance in vitro and in vivo. Neurochem. Int. 2001, 39, 435–448.
- Sharma, V.K.; Singh, T.G. Navigating Alzheimer’s Disease via Chronic Stress: The Role of Glucocorticoids. Curr. Drug Targets 2020, 21, 433–444.
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232.
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101.
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The Potential Role of the NLRP3 Inflammasome Activation as a Link between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front. Cell. Neurosci. 2019, 13, 73.
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954.
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 2017, 12, 7–12.
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583.
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting both BDNF/TrkB pathway and delta-secretase for treating Alzheimer’s disease. Neuropharmacology 2021, 197, 108737.
- Pak, M.E.; Yang, H.J.; Li, W.; Kim, J.K.; Go, Y. Yuk-Gunja-Tang attenuates neuronal death and memory impairment via ERK/CREB/BDNF signaling in the hippocampi of experimental Alzheimer’s disease model. Front. Pharmacol. 2022, 13, 1014840.
- Gupta, V.; Chitranshi, N.; Gupta, V.; You, Y.; Rajput, R.; Paulo, J.A.; Mirzaei, M.; van den Buuse, M.; Graham, S.L. TrkB Receptor Agonist 7,8 Dihydroxyflavone is Protective Against the Inner Retinal Deficits Induced by Experimental Glaucoma. Neuroscience 2022, 490, 36–48.
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer’s disease progression. Front. Mol. Neurosci. 2023, 16, 1150399.
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602.
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907.
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296.
- Kim, D.; Cho, J.; Kang, H. Protective effect of exercise training against the progression of Alzheimer’s disease in 3xTg-AD mice. Behav. Brain Res. 2019, 374, 112105.
- Xu, L.; Zhu, L.; Zhu, L.; Chen, D.; Cai, K.; Liu, Z.; Chen, A. Moderate Exercise Combined with Enriched Environment Enhances Learning and Memory through BDNF/TrkB Signaling Pathway in Rats. Int. J. Environ. Res. Public Health 2021, 18, 8283.
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692.
- Zhang, J.; Zheng, X.; Zhao, Z. A systematic review and meta-analysis on the efficacy outcomes of selective serotonin reuptake inhibitors in depression in Alzheimer’s disease. BMC Neurol. 2023, 23, 210.
- Casarotto, P.; Umemori, J.; Castrén, E. BDNF receptor TrkB as the mediator of the antidepressant drug action. Front. Mol. Neurosci. 2022, 15, 1032224.
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e1219.
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181.
- Håkansson, K.; Ledreux, A.; Daffner, K.; Terjestam, Y.; Bergman, P.; Carlsson, R.; Kivipelto, M.; Winblad, B.; Granholm, A.C.; Mohammed, A.K. BDNF Responses in Healthy Older Persons to 35 Minutes of Physical Exercise, Cognitive Training, and Mindfulness: Associations with Working Memory Function. J. Alzheimers Dis. 2017, 55, 645–657.
- Angelucci, F.; Peppe, A.; Carlesimo, G.A.; Serafini, F.; Zabberoni, S.; Barban, F.; Shofany, J.; Caltagirone, C.; Costa, A. A pilot study on the effect of cognitive training on BDNF serum levels in individuals with Parkinson’s disease. Front. Hum. Neurosci. 2015, 9, 130.
- Gomutbutra, P.; Srikamjak, T.; Sapinun, L.; Kunaphanh, S.; Yingchankul, N.; Apaijai, N.; Shinlapawittayatorn, K.; Phuackchantuck, R.; Chattipakorn, N.; Chattipakorn, S. Effect of intensive weekend mindfulness-based intervention on BDNF, mitochondria function, and anxiety. A randomized, crossover clinical trial. Compr. Psychoneuroendocrinol. 2022, 11, 100137.
- Canet, G.; Hernandez, C.; Zussy, C.; Chevallier, N.; Desrumaux, C.; Givalois, L. Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets. Front. Aging Neurosci. 2019, 11, 269.
- Watermeyer, T.; Robb, C.; Gregory, S.; Udeh-Momoh, C. Therapeutic implications of hypothalamic-pituitaryadrenal-axis modulation in Alzheimer’s disease: A narrative review of pharmacological and lifestyle interventions. Front. Neuroendocrinol. 2021, 60, 100877.
- Sooy, K.; Webster, S.P.; Noble, J.; Binnie, M.; Walker, B.R.; Seckl, J.R.; Yau, J.L. Partial deficiency or short-term inhibition of 11beta-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J. Neurosci. 2010, 30, 13867–13872.
- Sooy, K.; Noble, J.; McBride, A.; Binnie, M.; Yau, J.L.; Seckl, J.R.; Walker, B.R.; Webster, S.P. Cognitive and Disease-Modifying Effects of 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibition in Male Tg2576 Mice, a Model of Alzheimer’s Disease. Endocrinology 2015, 156, 4592–4603.
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 2013, 74, 357–366.
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol. Aging 2016, 45, 109–122.
- da Costa Daniele, T.M.; de Bruin, P.F.C.; de Matos, R.S.; de Bruin, G.S.; Maia Chaves, C.J.; de Bruin, V.M.S. Exercise effects on brain and behavior in healthy mice, Alzheimer’s disease and Parkinson’s disease model-A systematic review and meta-analysis. Behav. Brain Res. 2020, 383, 112488.
- Bashiri, H.; Enayati, M.; Bashiri, A.; Salari, A.A. Swimming exercise improves cognitive and behavioral disorders in male NMRI mice with sporadic Alzheimer-like disease. Physiol. Behav. 2020, 223, 113003.
- Campos, H.C.; Ribeiro, D.E.; Hashiguchi, D.; Glaser, T.; Milanis, M.D.S.; Gimenes, C.; Suchecki, D.; Arida, R.M.; Ulrich, H.; Monteiro Longo, B. Neuroprotective effects of resistance physical exercise on the APP/PS1 mouse model of Alzheimer’s disease. Front. Neurosci. 2023, 17, 1132825.
- Irazoki, E.; Contreras-Somoza, L.M.; Toribio-Guzmán, J.M.; Jenaro-Río, C.; van der Roest, H.; Franco-Martín, M.A. Technologies for Cognitive Training and Cognitive Rehabilitation for People With Mild Cognitive Impairment and Dementia. A Systematic Review. Front. Psychol. 2020, 11, 648.
- Cutuli, D.; Decandia, D.; Giacovazzo, G.; Coccurello, R. Physical Exercise as Disease-Modifying Alternative against Alzheimer’s Disease: A Gut-Muscle-Brain Partnership. Int. J. Mol. Sci. 2023, 24, 4686.