 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pietro Traldi | -- | 4677 | 2024-02-03 10:08:38 | | | |
| 2 | Lindsay Dong | + 2 word(s) | 4679 | 2024-02-04 02:28:36 | | |
Video Upload Options
Drug resistance remains one of the main causes of poor outcome in cancer therapy. It is also becoming evident that drug resistance to both chemotherapy and to antibiotics is driven by more than one mechanism. So far, there are at least eight recognized mechanisms behind such resistance. In normal tissues, ATP-binding cassette (ABC) transporters protect the cells from the toxic effects of xenobiotics, whereas in tumor cells, they reduce the intracellular concentrations of anticancer drugs, which ultimately leads to the emergence of multidrug resistance (MDR). A deeper understanding of the structures and the biology of these proteins is central to current efforts to circumvent resistance to both chemotherapy, targeted therapy, and antibiotics.
1. Introduction
1.1. Developments Which Enhanced the Role of MS in Proteins Characterization
1.1.1. Hydrogen–Deuterium Exchange Mass Spectrometry(H/DX-MS)
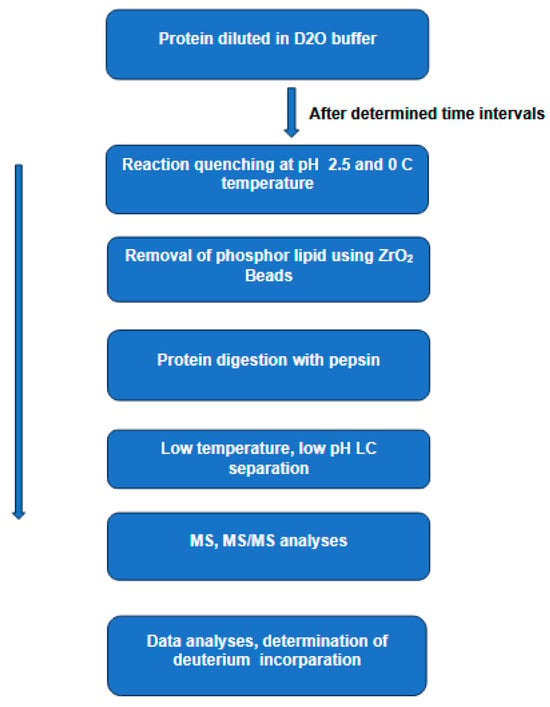
1.1.2. Ion Mobility–Mass Spectrometry
1.1.3. More Efficient Activation Methods of Macromolecules
1.1.4. Electron Capture Dissociation (ECD)
1.1.5. Electron Transfer Dissociation (ETD)
1.1.6. Photodissociation Methods
2. Analysis of Some ATP-Binding Cassette (ABC) Transporters
The introduction of two soft ionization techniques, namely electrospray ionization (ESI) [64] and matrix-assisted laser desorption/ionization (MALDI) [65], was a major step towards a diffused use of mass spectrometry (MS) in biological and biochemical worlds. Over the last two decades, advances in MS instrumentation together with more refined labelling protocols, the combination of ion mobility with MS, and more frequent use of electron-based and photon-based fragmentation methods paved the way for what is known today as structural mass spectrometry. With these innovative developments, mass spectrometry can be applied for the structural characterization of a wide range of macromolecules and in some cases their respective assemblies. It is relevant to point out that continuous progress in sample preparations, including the use of a new class of detergents, extended the application of MS to membrane proteins, which are not water soluble.
2.1. Monitoring the Conformation of P-glycoprotein
2.2. Breast Cancer Resistance Protein (ABCG2)
3. Commenting on Inhibitors of P-glycoprotein
4. Conclusions
References
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50.
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038.
- Arinaminpathy, Y.; Khurana, E.; Engelman, D.M.; Gerstein, M.B. Computational analysis of membrane proteins: The largest class of drug targets. Drug Discov. Today 2009, 14, 1130–1135.
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. Opinion—How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996.
- Billings, E.; Lundquist, K.; Overly, C.; Srinivasan, K.; Noinaj, N. Structure Determination of Membrane Proteins Using X-ray Crystallography. Methods Mol. Biol. 2021, 2302, 101–136.
- Hernando-Amado, S.; Blanco, P.; Alcalde-Rico, M.; Corona, F.; Reales-Calderón, J.A.; Sánchez, M.B.; Martínez, J.L. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist. Update 2016, 28, 13–27.
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267.
- Levin, B.R.; Rozen, D.E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006, 4, 556–562.
- Corona, F.; Martinez, J.L. Phenotypic Resistance to Antibiotics. Antibiotics 2013, 2, 237–255.
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58.
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398.
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30.
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165.
- Kühlbrandt, W. The Resolution Revolution. Science 2014, 343, 1443–1444.
- Thompson, R.F.; Walker, M.; Siebert, C.A.; Muench, S.P.; Ranson, N.A. An introduction to sample preparation and imaging by cryo-electron microscopy for structural biology. Methods 2016, 100, 3–15.
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234.
- Calabrese, A.N.; Radford, S.E. Mass spectrometry-enabled structural biology of membrane proteins. Methods 2018, 147, 187–205.
- Laganowsky, A.; Reading, E.; Hopper, J.T.S.; Robinson, C.V. Mass spectrometry of intact membrane protein complexes. Nat. Protoc. 2013, 8, 639–651.
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294.
- Konijnenberg, A.; Butterer, A.; Sobott, F. Native ion mobility-mass spectrometry and related methods in structural biology. BBA—Proteins Proteom. 2013, 1834, 1239–1256.
- Zhong, Y.; Hyung, S.-J.; Ruotolo, B.T. Ion mobility–mass spectrometry for structural proteomics. Expert Rev. Proteom. 2012, 9, 47–58.
- Engen, J.R.; Wales, T.E. Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu. Rev. Anal. Chem. 2015, 8, 127–148.
- Katta, W.; Chait, B.T. Hydrogen/Deuterium Exchange Electrospray Ionization Mass Spectrometry: A Method for Probing Protein Conformational Changes in Solution. J. Am. Chem. Soc. 1993, 115, 6317–6321.
- Hammerschmid, D.; Calvaresi, V.; Bailey, C.; Russell, B.; Argyris Politis, L.; Morris, M.; Denbigh, L.; Anderson, M.; Reading, E. Chromatographic Phospholipid Trapping for Automated H/D Exchange Mass Spectrometry of Membrane Protein–Lipid assemblies. Anal. Chem. 2023, 95, 3002–3011.
- Calvaresi, V.; Redsted, A.; Norais, N.; Rand, K.D. Hydrogen–Deuterium exchange mass spectrometry with integrated size-exclusion chromatography for analysis of complex protein samples. Anal. Chem. 2021, 93, 11406–11414.
- Donnarumma, D.; Maestri, C.; Giammarinaro, P.I.; Capriotti, L.; Bartolini, E.; Veggi, D.; Petracca, R.; Scarselli, M.; Norais, N. Native state organization of outer membrane porins unraveled by HDx-MS. J. Proteome Res. 2018, 17, 1794–1800.
- Mysling, S.; Salbo, R.; Ploug, M.; Jørgensen, T.J.D. Electrochemical reduction of disulfide-containing proteins for hydrogen/deuterium exchange monitored by mass spectrometry. Anal. Chem. 2014, 86, 340–345.
- Bobst, C.E.; Kaltashov, I.A. Enhancing the Quality of H/D Exchange measurements with mass spectrometry detection in disulfide-rich proteins using electron capture dissociation. Anal. Chem. 2014, 86, 5225–5231.
- Burns, J.A.; Butler, J.C.; Moran, J.; Whitesides, G.M. Selective reduction of disulfides by tris(2-carboxyethyl)phosphine. J. Org. Chem. 1991, 56, 2648–2650.
- Larsen, M.R.; Højrup, P.; Roepstorff, P. Characterization of Gel-separated Glycoproteins Using Two-step Proteolytic Digestion Combined with Sequential Microcolumns and Mass Spectrometry. Mol. Cell. Proteom. 2005, 4, 107–119.
- Darula, Z.; Medzihradszky, K.F. Glycan side reaction may compromise ETD-based glycopeptide identification. J. Am. Soc. Mass Spectrom. 2014, 25, 977–987.
- Houel, S.; Hilliard, M.; Yu, Y.Q.; McLoughlin, N.; Martin, S.M.; Rudd, P.M.; Williams, J.P.; Chen, W. N- and O-glycosylation analysis of etanercept using liquid chromatography and quadrupole time-of-flight mass spectrometry equipped with electron-transfer dissociation functionality. Anal. Chem. 2014, 86, 576–584.
- Rand, K.D. Pinpointing changes in higher-order protein structure by hydrogen/deuterium exchange coupled to electron transfer dissociation mass spectrometry. Int. J. Mass Spectrom. 2013, 338, 2–10.
- Javed, W.; Griffiths, D.; Politis, A. Hydrogen/deuterium exchange-mass spectrometry of integral membrane proteins in native-like environments: Current scenario and the way forward. Essays Biochem. 2023, 67, 187–200.
- Majeed, S.; Ahmad, A.B.; Sehar, U.; Georgieva, E.R. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes 2021, 11, 685.
- Karasek, F.W. Drift-mass spectrometer. Res. Dev. 1970, 21, 25–27.
- Alge, E.; Villinger, H.; Lindinger, W. Drift tube investigations on the reactions of O2 + with CH4 and of CO2 + with NO in various buffer gases. Plasma Chem. Plasma Process. 1981, 1, 65–71.
- Smith, D.P.; Giles, K.; Bateman, R.H.; Radford, S.E.; Ashcroft, A.E. Monitoring Copopulated Conformational States during Protein Folding Events Using Electrospray Ionization-Ion Mobility Spectrometry-Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2007, 18, 2180–2190.
- May, J.C.; Morris, C.B.; McLean, J.A. Ion mobility collision cross section compendium. Anal. Chem. 2017, 89, 1032–1044.
- Wyttenbach, T.; Bowers, M.T. Structural stability from solution to the gas phase: Native solution structure of ubiquitin survives analysis in a solvent-free ion mobility–mass spectrometry environment. J. Phys. Chem. B 2011, 115, 12266–12275.
- Ruotolo, B.T.; Robinson, C.V. Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 2006, 10, 402–408.
- Yost, R.A.; Enke, C.G. Selected ion fragmentation with a tandem quadrupole mass spectrometer. J. Am. Chem. Soc. 1978, 100, 2274–2275.
- Zubarev, R.A.; Horn, D.M.; Fridriksson, E.K.; Kelleher, N.L.; Kruger, N.A.; Lewis, M.A.; Carpenter, B.K.; McLafferty, F.W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000, 72, 563–573.
- Syrstad, E.A.; Turecček, F. Toward a general mechanism of electron capture dissociation. J. Am. Soc. Mass Spectrom. 2005, 16, 208–224.
- Zubarev, R.A.; Kelleher, N.L.; McLafferty, F.W. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 1998, 120, 3265–3266.
- Qi, Y.; Volmer, D.A. Electron-based fragmentation methods in mass spectrometry: An overview. Mass Spectrom. Rev. 2017, 36, 4–15.
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and Protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533.
- Pitteri, S.J.; Chrisman, P.A.; McLuckey, S.A. Electron-Transfer Ion/Ion Reactions of Doubly Protonated Peptides: Effect of Elevated Bath Gas Temperature. Anal. Chem. 2005, 77, 5662–5669.
- Ko, B.J.; Brodbelt, J.S. Enhanced electron transfer dissociation of peptides modified at C-terminus with fixed charges. J. Am. Soc. Mass Spectrum. 2012, 23, 1991–2000.
- Frey, B.L.; Ladror, D.T.; Sondalle, S.B.; Krusemark, C.J.; Jue, A.L.; Coon, J.J.; Smith, L.M. Chemical derivatization of peptide carboxyl groups for highly efficient electron transfer dissociation. J. Am. Soc. Mass Spectrom. 2013, 24, 1710–1721.
- Swaney, D.L.; McAlister, G.C.; Coon, J.J. Decision tree–driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 2008, 5, 959–964.
- Frese, C.K.; Altelaar, A.F.M.; Hennrich, M.L.; Nolting, D.; Zeller, M.; Griep-Raming, J.; Heck, A.J.R.; Mohammed, S. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-orbitrap velos. J. Proteome Res. 2011, 10, 2377–2388.
- Woodin, R.L.; Bomse, D.S.; Beauchamp, J.L. Multiphoton dissociation of molecules with low power continuous wave infrared laser radiation. J. Am. Chem. Soc. 1978, 100, 3248–3250.
- Maitre, P.; Scuderi, D.; Corinti, D.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Applications of Infrared Multiple Photon Dissociation (IRMPD) to the Detection of Posttranslational Modifications. Chem. Rev. 2020, 120, 3261–3295.
- Greisch, J.-F.; van der Laarse, S.A.; Heck, A.J. Enhancing Top-Down Analysis Using Chromophore-Assisted Infrared Multiphoton Dissociation from (Phospho)peptides to Protein Assemblies. Anal. Chem. 2020, 92, 15506–15516.
- Bowers, W.D.; Delbert, S.S.; Hunter, R.L.; Mciver, R.T. Fragmentation of oligopeptide ions using ultraviolet laser radiation and fourier transform mass spectrometry. J. Am. Chem. Soc. 1984, 106, 7288–7289.
- Hunt, D.F.; Shabanowitz, J.; Yates, J.R. Peptide sequence analysis by laser photodissociation Fourier transform mass spectrometry. J. Chem. Soc. Chem. Commun. 1987, 548–550.
- Thompson, M.S.; Cui, W.; Reilly, J.P. Fragmentation of Singly Charged Peptide Ions by Photodissociation at λ = 157 nm. Angew. Chem. Int. Ed. 2004, 43, 4791–4794.
- Cannon, J.R.; Cammarata, M.B.; Robotham, S.A.; Cotham, V.C.; Shaw, J.B.; Fellers, R.T.; Early, B.P.; Thomas, P.M.; Kelleher, N.L.; Brodbelt, J.S. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal. Chem. 2014, 86, 2185–2192.
- Smyrnakis, A.; Levin, N.; Kosmopoulou, M.; Jha, A.; Fort, K.; Makarov, A.; Papanastasiou, D.; Mohammed, S. Characterization of an Omnitrap-Orbitrap Platform Equipped with Infrared Multiphoton Dissociation, Ultraviolet Photodissociation, and Electron Capture Dissociation for the Analysis of Peptides and Proteins. Anal. Chem. 2023, 95, 12039–12046.
- Fornelli, L.; Srzentić, K.; Toby, T.K.; Doubleday, P.F.; Huguet, R.; Mullen, C.; Melani, R.D.; dos Santos Seckler, H.; DeHart, C.J.; Weisbrod, C.R.; et al. Thorough Performance Evaluation of 213 nm Ultraviolet Photodissociation for Top-down Proteomics. Mol. Cell. Proteom. 2020, 19, 405–420.
- Julian, R.R.; Amster, I.J.; Kong, X.; Brodbelt, J.S.; Jørgensen, T.J.D.; Wysocki, V.H.; Hendrickson, C.L.; Santos, I.; Shaw, J.B.; Boyarkin, O.V.; et al. The mechanism behind top-down uvpd experiments: Making sense of apparent contradictions. J. Am. Soc. Mass Spectrom. 2017, 28, 1823–1826.
- Zabuga, A.V.; Kamrath, M.Z.; Boyarkin, O.V.; Rizzo, T.R. Fragmentation mechanism of UV-excited peptides in the gas phase. J. Chem. Phys. 2014, 141, 154309.
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459.
- Karas, M.; Bachmann, D.; Hillenkamp, F. Influence of the Wavelength in High-Irradiance Ultraviolet Laser Desorption Mass Spectrometry of Organic Molecules. Anal. Chem. 1985, 57, 2935–2939.
- Juliano, R.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. BBA—Biomembr. 1976, 455, 152–162.
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.-W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391.
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29.
- Marquez, B.; Van Bambeke, F. ABC multidrug transporters: Target for modulation of drug pharmacokinetics and drug-drug interactions. Curr. Drug Targets 2011, 12, 600–620.
- Kopcho, N.; Geoffrey Chang, G.; Komives, E.A. Dynamics of ABC transporter P-glycoprotein in three conformational states. Sci. Rep. 2019, 9, 15092.
- Van Herwaarden, A.E.; Schinkel, A.H. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol. Sci. 2006, 27, 10–16.
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509.
- Khunweeraphong, N.; Stockner, T.; Kuchler, K. The structure of the human ABC transporter ABCG2 reveals a novel mechanism for drug extrusion. Sci. Rep. 2017, 7, 13767.
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340.
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430.
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. 1998, 95, 15665–15670.
- Zhang, Y.; Vagiannis, D.; Budagaga, Y.; Sabet, Z.; Hanke, I.; Rozkoš, T.; Hofman, J. Sonidegib potentiates the cancer cells’ sensitivity to cytostatic agents by functional inhibition of ABCB1 and ABCG2 in vitro and ex vivo. Biochem. Pharmacol. 2022, 199, 115009.
- Modok, S.; Mellor, H.R.; Callaghan, R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr. Opin. Pharmacol. 2006, 6, 350–354.
- Crowley, E.; McDevitt, C.A.; Callaghan, R. Generating Inhibitors of P-Glycoprotein: Where to, now? Multi-Drug Resist. Cancer 2010, 596, 405–432.

