Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina M. Caruso | -- | 3766 | 2024-01-31 17:53:29 | | | |
| 2 | Jason Zhu | Meta information modification | 3766 | 2024-02-01 02:23:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Padovani, C.M.; Yin, K. Mechanisms of Immunosuppression in Sepsis. Encyclopedia. Available online: https://encyclopedia.pub/entry/54611 (accessed on 26 June 2026).
Padovani CM, Yin K. Mechanisms of Immunosuppression in Sepsis. Encyclopedia. Available at: https://encyclopedia.pub/entry/54611. Accessed June 26, 2026.
Padovani, Cristina M., Kingsley Yin. "Mechanisms of Immunosuppression in Sepsis" Encyclopedia, https://encyclopedia.pub/entry/54611 (accessed June 26, 2026).
Padovani, C.M., & Yin, K. (2024, January 31). Mechanisms of Immunosuppression in Sepsis. In Encyclopedia. https://encyclopedia.pub/entry/54611
Padovani, Cristina M. and Kingsley Yin. "Mechanisms of Immunosuppression in Sepsis." Encyclopedia. Web. 31 January, 2024.
Copy Citation
Severe infection can lead to sepsis. In sepsis, the host mounts an inappropriately large inflammatory response in an attempt to clear the invading pathogen. This sustained high level of inflammation may cause tissue injury and organ failure. Later in sepsis, a paradoxical immunosuppression occurs, where the host is unable to clear the preexisting infection and is susceptible to secondary infections. A major issue with sepsis treatment is that it is difficult for physicians to ascertain which stage of sepsis the patient is in. Sepsis treatment will depend on the patient’s immune status across the spectrum of the disease, and these immune statuses are nearly polar opposites in the early and late stages of sepsis.
macrophages
lymphocytes
apoptosis
infection
MDSCs
1. Monocyte/Macrophage Exhaustion
One hallmark of sepsis-induced immunosuppression is monocyte/macrophage exhaustion [1]. Monocytes are circulating white blood cells derived from the bone marrow and are major players in the innate immune system. Their primary role is to detect changes in homeostasis (usually caused by an infection and/or tissue damage) and respond to these changes by replenishing the pool of macrophages that are present in the tissues [2]. Once monocytes become activated, they turn into effector cells and produce cytokines, present antigens to adaptive immune cells, phagocytose, efferocytose, initiate inflammation, or resolve inflammation [2]. When monocytes initiate tissue inflammation, they exit the blood and enter the tissues at the site of the inflammation, thereby becoming macrophages. Macrophages can either be recruited to a specific tissue (which is what happens during the inflammatory response), or they can exist as residents within the body’s tissues. Macrophages can polarize into different subtypes depending on which subtype can best assist the host at that time. The previously accepted polarization classes are M0, M1-like, and M2-like; an M0 macrophage is a nonactivated tissue resident macrophage, an M1-like macrophage is a pro-inflammatory macrophage, and an M2-like macrophage is an anti-inflammatory macrophage (See Figure 1) [2]. It is classically understood that M1-like macrophages have increased production of pro-inflammatory cytokines, enhanced antigen presentation, and enhanced phagocytosis capabilities [3]. M2-like macrophages have diminished pro-inflammatory cytokine output and weakened antigen presentation, but enhanced phagocytosis capabilities [3][4]. These phenotypes can be artificially induced in vitro via monocyte stimulation, with LPS and IFN-γ leading to the development of a “classic” M1-like macrophage and IL-4 leading to the development of a “classic” M2-like macrophage [5][6].
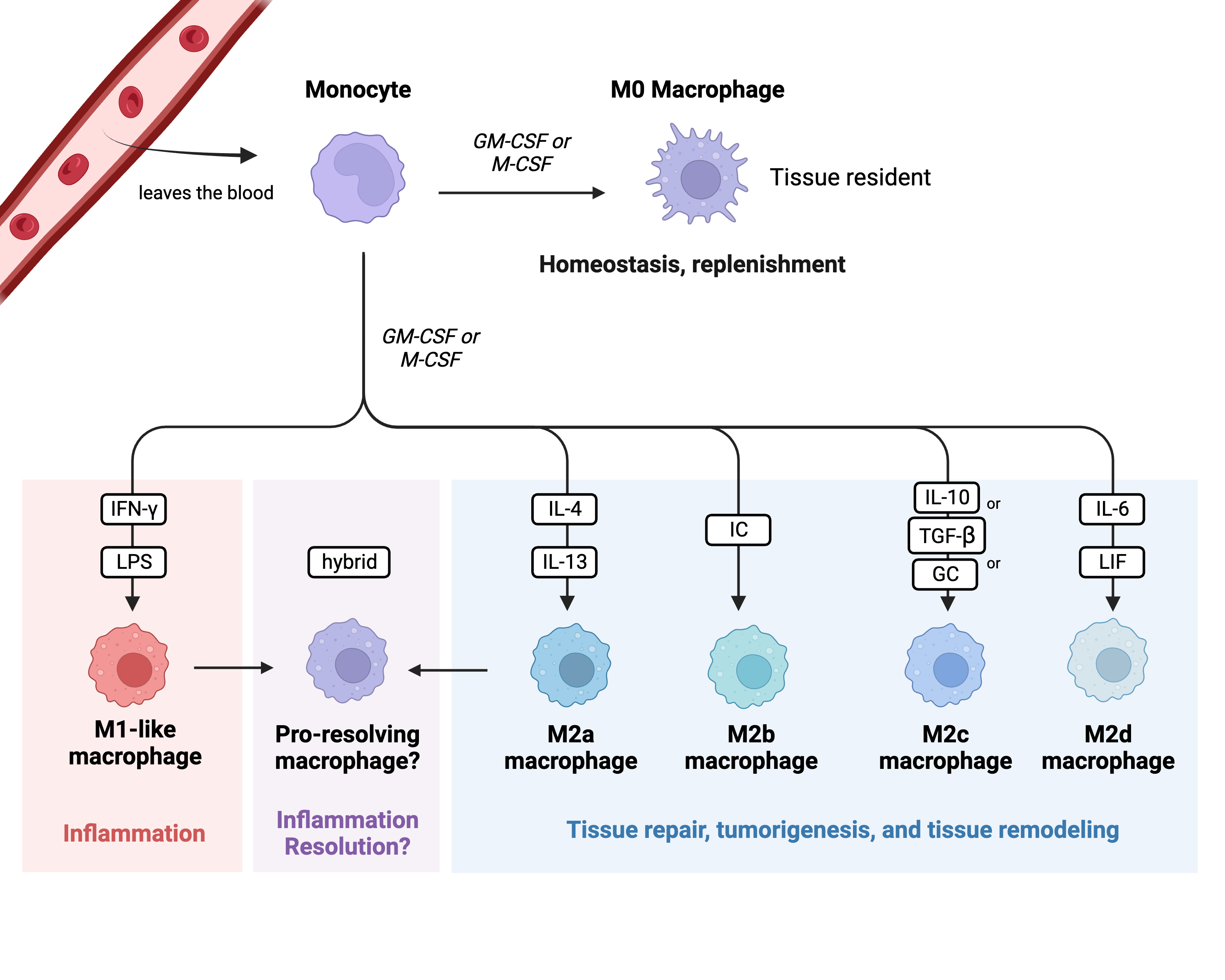
Figure 1. Monocyte polarization. This figure highlights the classical monocyte polarization scheme and classes of macrophages. Each class has a unique role within the immune system. Key: GM-CSF: granulocyte macrophage colony-stimulating factor; M-CSF: macrophage colony-stimulating factor; LPS: lipopolysaccharide; IC: immune complex; GC: glucocorticoid; LIF: leukemia inhibitory factor. Created in BioRender.com.
M1-like macrophages are stimulated by IFN-γ and LPS; they are key initiators of the inflammatory response and are critical for the removal of bacteria [7]. On the other hand, M2-like macrophages are highly phagocytic and are key drivers of tissue remodeling following damage, tumor progression via the production of angiogenic factors, and parasite removal [7]. M2-like macrophages are also a main driver in the development of allergies. Additionally, the M2-like macrophage subtype can be further subclassified into M2a, M2b, M2c, and M2d [8][9]. These subtypes vary in what cytokines they release, what surface markers they express, and what specific roles they play within M2-like macrophage responses. M2a macrophages are stimulated via IL-4 and IL-13; they enhance cell growth and tissue repair and enhance macrophage endocytosis [8][9]. M2b macrophages are stimulated via immune complexes (IC); they are able to regulate the host’s immune status by secreting both pro- and anti-inflammatory cytokines [8][9]. M2b macrophages are also well known for their ability to promote tumor progression [9]. M2c macrophages are stimulated by IL-10, TGF-β, or glucocorticoids (GCs); like M2b macrophages, they are highly immune-regulatory but specialize in efferocytosis and phagocytosis of other apoptotic cells [8][9]. M2d macrophages are stimulated via IL-6 and leukemia inhibitory factor (LIF); they promote angiogenesis and tumor progression via the release of vascular endothelial growth factors and dampening of the immune response [8][9].
While the M1/M2 paradigm is helpful for investigators studying these different capabilities of macrophages in vitro, many researchers find that limiting the description of macrophage phenotypes to only M1-like or M2-like is not clinically relevant, as this tidy polarization process does not occur at a tissue level [5][6][10][11][12]. Current research explains that activated macrophages have high levels of plasticity and do not solely polarize into an M1-like or M2-like phenotype at the tissue level; rather, they adopt a phenotype that is highly dependent on both the environment they are in and their own ontogeny [5][6]. One such newly reported phenotype is the “pro-resolving” macrophage phenotype [5][6][13][14]. These dynamic, pro-resolving macrophages function primarily to resolve inflammation so that the host can return to homeostasis, while simultaneously contributing to wound/tissue repair [5][6]. Interestingly, these pro-resolving macrophages are said to have qualities of the classic M1- and M2-like phenotypes, with aspects that contribute to both pro-inflammatory and anti-inflammatory pathways [13]. Some studies even report that abnormal activation of this pro-resolving phenotype can lead to chronic organ dysfunction, highlighting their direct importance in the inflammation resolution process [15][16][17].
1.1. PD-1 and PDL-1
Monocytes/macrophages contribute to sepsis-induced immunosuppression in a number of ways. Monocytes/macrophages can modulate the expression of their immune regulatory markers, an action that has implications for other immune cells and physiological pathways [18]. Changes in the expression of immune regulatory markers such as CTLA-4, PD-1, and PD-L1 can alter these interactions [19]. PD-1 (also referred to as CD279) is a transmembrane protein located on the surfaces of many immune cells; it is expressed in the highest concentration on activated T cells [20]. When PD-1 interacts with its ligand PD-L1, the T cell becomes anergic, or unresponsive to viable antigens [21][22][23][24]. PD-L1 is expressed by certain APCs (such as monocytes/macrophages) in inflammatory scenarios as well as by tumor cells [21]. Additionally, certain pro-inflammatory cytokines (especially IFN-γ) can trigger various signal transduction cascades within APCs, leading directly to increased expression of PD-L1 on their surfaces [25].
Physiologically, the PD-1/PD-L1 pathway aids in controlling the degree of inflammation at the tissue level by deactivating T cells that are directly contributing to the inflammation so as to protect tissues from immune-mediated damage [21]. Tumor cells overexpress PD-L1 in order to exist for longer periods of time without being “seen” by the adaptive immune system [21][25]. The PD-1/PD-L1 pathway has therefore become a major contributor to immunosuppression within the tumor microenvironment in many different types of cancers (notably melanoma and small-cell lung cancer) [26]. Understanding this major contribution of PD-1/PD-L1 to cancer-induced immunosuppression has led to the development of a new class of cancer immunotherapies called checkpoint inhibitors [19][26]. Due to many parallels existing between cancer-induced immunosuppression and sepsis-induced immunosuppression [1], checkpoint inhibitors were tested in clinical trials of sepsis patients and cecal ligation and puncture (CLP) sepsis models [27][28]. Hotchkiss et al. was the first to study the effects and safety profile of the anti-PD-1 immunotherapy agent nivolumab in a small clinical trial of sepsis patients [27]. More recently, Yang et al. showed that blocking PD-L1 on monocytes led to a dramatic increase in survival of CLP-septic mice [28].
Additionally, many researchers have assessed the potential correlation between immune regulatory marker expression and outcomes due to sepsis. Ruan et al. found that there is an increased number of cells expressing both PD-1 and PD-L1 in CLP-septic mice [29]. This finding was also true for humans, as there are reported increases in expression of both PD-1 and PD-L1 in human septic patients [29][30][31][32]. While changes in immune regulatory marker expression on monocytes/macrophages contributes to sepsis-induced immunosuppression, this finding is not specific to septic patients. As previously mentioned, cancer cells can implement similar changes in regulatory marker expression levels in order to evade the immune system. A more explicit example of monocytes/macrophages contributing to immunosuppression due to sepsis is monocyte/macrophage exhaustion [1].
1.2. Exhausted Monocytes/Macrophages
Exhausted monocytes/macrophages are “hypoactive”, meaning that they have a decreased capacity to perform their normal effector functions when stimulated [33][34][35][36]. These normal effector functions include presenting antigens via HLA-DR, phagocytosing, efferocytosing, and releasing pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-8) via the NF-κB pathway [2]. Therefore, NF-κB status is an accepted marker of exhaustion [37]. As monocytes/macrophages from septic patients also have similarly reduced capacities of these normal functions, it can be inferred that septic monocyte. The authors found that whole blood samples taken from 61 septic patients (blood taken within 24 h of meeting sepsis-3 criteria) had an overwhelming presence of inflammation, yet leukocytes from these samples had depressed responses to both LPS and bacteria [38], providing evidence that septic leukocytes were similar to the “exhausted” phenotype. This hypoactive phenotype present in septic monocytes/macrophages can be recapitulated in vitro by repetitively challenging monocytes/macrophages with isolated, low-dose bacterial endotoxins such as LPS [35][39].
It is important to briefly discuss here that monocyte/macrophage exhaustion is different from the phenomenon called endotoxin tolerance (ET). ET has many similar characteristics to exhaustion [40], and it is defined as reduced immune cell functional responsiveness to the bacterial endotoxin LPS after previously encountering LPS [33][34]. Blackwell et al. examined ET via monocyte cytokine production; they discovered that tolerized monocytes had impaired activation of NF-κB [41], which is also observed in exhaustion. Tolerized monocytes/macrophages exhibited a reduced capacity to react to subsequent LPS challenge; primarily of note was the drastic decrease in NF-κB-mediated TNF-α production upon LPS restimulation [42][43][44][45][46][47]. While the monocytes from late-septic patients seem to have many aspects similar to monocytes/macrophages experiencing ET, the conventional understanding of ET does not encompass the fundamental characteristics of immunosuppression observed in septic patients [36]. For example, monocytes/macrophages experiencing ET have enhanced bacterial phagocytosis, as it has been theorized that ET monocytes/macrophages adopt a phenotype similar to M2-like macrophages [33]. However, exhausted monocytes/macrophages and monocytes from septic patients demonstrate impaired bacterial phagocytosis, even though the mechanism underlying this decline is not known [48][49]. For these reasons, the emerging literature more accurately describes septic monocytes/macrophages as exhausted rather than tolerized [36]. However, the mechanism(s) underlying monocyte/macrophage exhaustion during the immunosuppressive stage of sepsis remains elusive.
Several possibilities for sepsis-induced monocyte/macrophage exhaustion include increased production of the anti-inflammatory cytokine IL-10, down-regulation of the NF-κB signaling pathway, decreased HLA-DR expression, and reduced lymphocyte signaling due to lymphocyte cell death [34][40][47]. Another possibility is the high mobility group box 1 (HMGB1) protein, which has been shown to increase macrophage pyroptosis [50][51][52], decrease macrophage efferocytosis ability [53], and enhance the suppressive activity of an immunosuppressive cell population called myeloid-derived suppressor cells (MDSCs) [54].
To overcome monocyte/macrophage anergy (i.e., exhaustion) several research teams have used IFN-γ in order to stimulate monocyte activity [55]. In one study performed by Döcke et al., the authors were able to increase HLA-DR expression in anergic monocytes, leading to sepsis resolution in eight of nine septic patients [55]. On the other hand, a more recent study showed that increased plasma IFN-γ levels were associated with secondary Candida sp. infections in late-sepsis patients [56]. Surprisingly, the authors also showed that IFN-γ reduced macrophage phagocytosis of zymosan particles [56]. Together, these two contradictory studies suggest that IFN-γ cannot be reliably regarded as an appropriate treatment to reverse sepsis-induced monocyte/macrophage exhaustion.
2. Lymphocyte Apoptosis
Another hallmark of sepsis-induced immunosuppression is lymphocyte apoptosis [1]. Before apoptosis can be discussed in more detail, it is important to mention that there are numerous types of regulated and unregulated cell death processes involving complex signaling cascades. The regulated and clinically relevant processes include, but are not limited to, autophagy, ferroptosis, pyroptosis, necroptosis, and apoptosis (See Figure 2). Autophagy is a process of regulated cell death by which autophagosomes collect cell waste products and deliver them to lysosomes for destruction [57]. This process is mainly employed to maintain homeostasis within the cell and to manage lipid metabolism [58]. Ferroptosis is a process of regulated cell death and is primarily caused by the accumulation of iron [59]. This accumulation leads to the development of reactive oxygen species (ROS), which contributes to both cell membrane rupture and lipid peroxidation [58]. Pyroptosis is a process of regulated cell death whereby destruction of the cell membrane is triggered by the activation of a multi-protein complex called the inflammasome [59][60]. Necroptosis is a process of regulated cell death that eventually leads to an influx of ions, swelling of the cell, and eventual breakdown of the membrane [59]. Due to many similarities between necroptosis and apoptosis (apoptosis is described in detail below), it has been suggested that necroptosis exists as a backup mechanism for apoptosis [58].
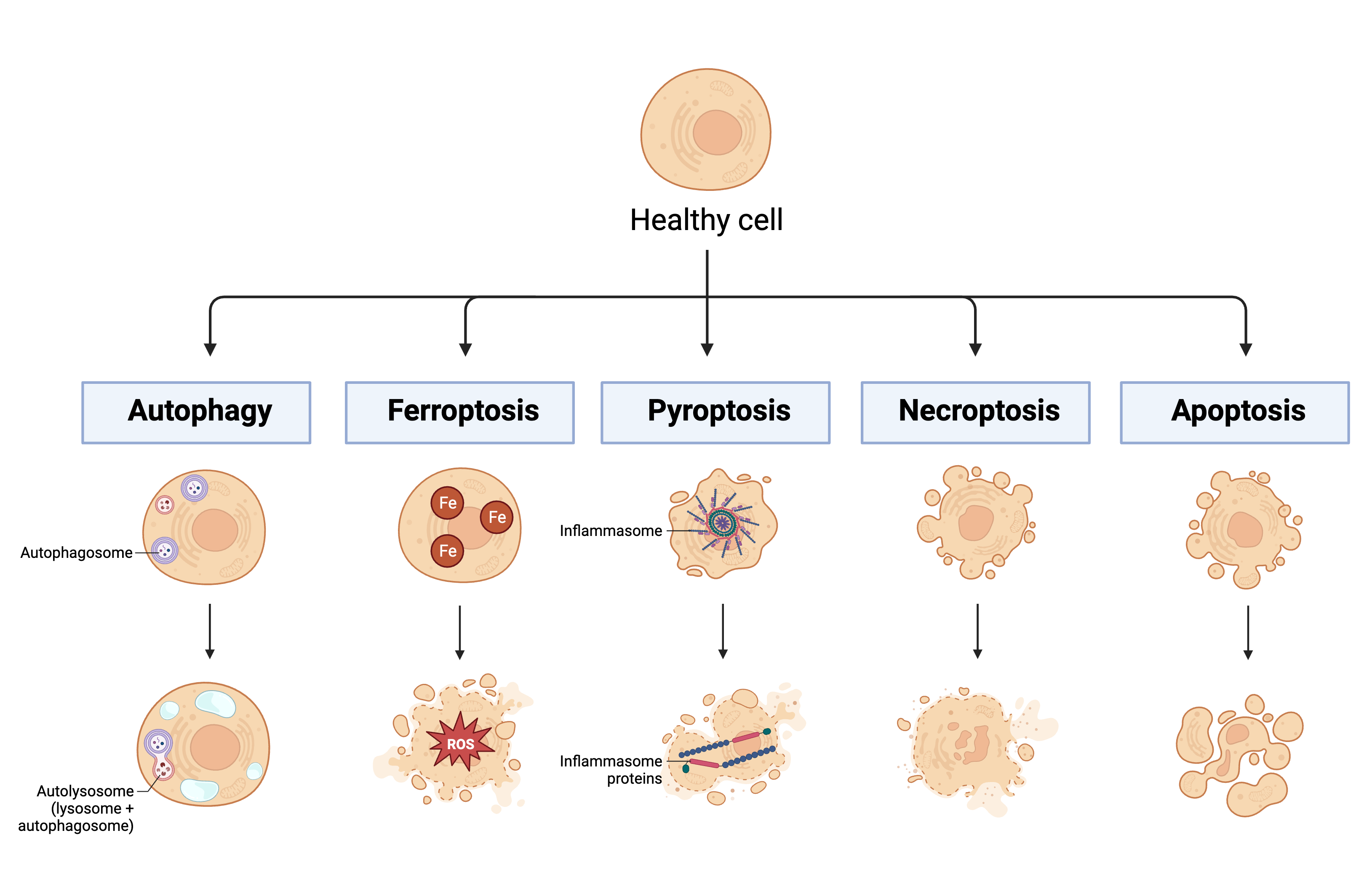
Figure 2. Mechanisms of regulated cell death. This figure highlights some important processes of regulated cell death: autophagy, ferroptosis, pyroptosis, necroptosis, and apoptosis. Key: Fe: iron; ROS: reactive oxygen species. Created in BioRender.com.
Apoptosis, or programmed cell death, is an active cellular process that is essential for proper functioning and regulation of many cells within the body, including immune cells. Apoptosis of faulty lymphocytes during and following lymphocyte development is crucial for proper immune functioning. There are two pathways that lead to lymphocyte apoptosis: “death by neglect” and “death by instruction” (See Figure 3) [61]. As discussed earlier, there are many mechanisms in place during lymphocyte development and activation to ensure proper performance and homeostasis within the population. If a lymphocyte fails to meet certain functional requirements, it will die via apoptosis (“death by neglect”). On the other hand, if a lymphocyte functions too aggressively, it will die via apoptosis (“death by instruction”). These apoptosis pathways that regulate lymphocyte development and expansion are necessary to ensure there are no self-reactive and/or hyporesponsive lymphocytes present in the immune system’s repertoire.
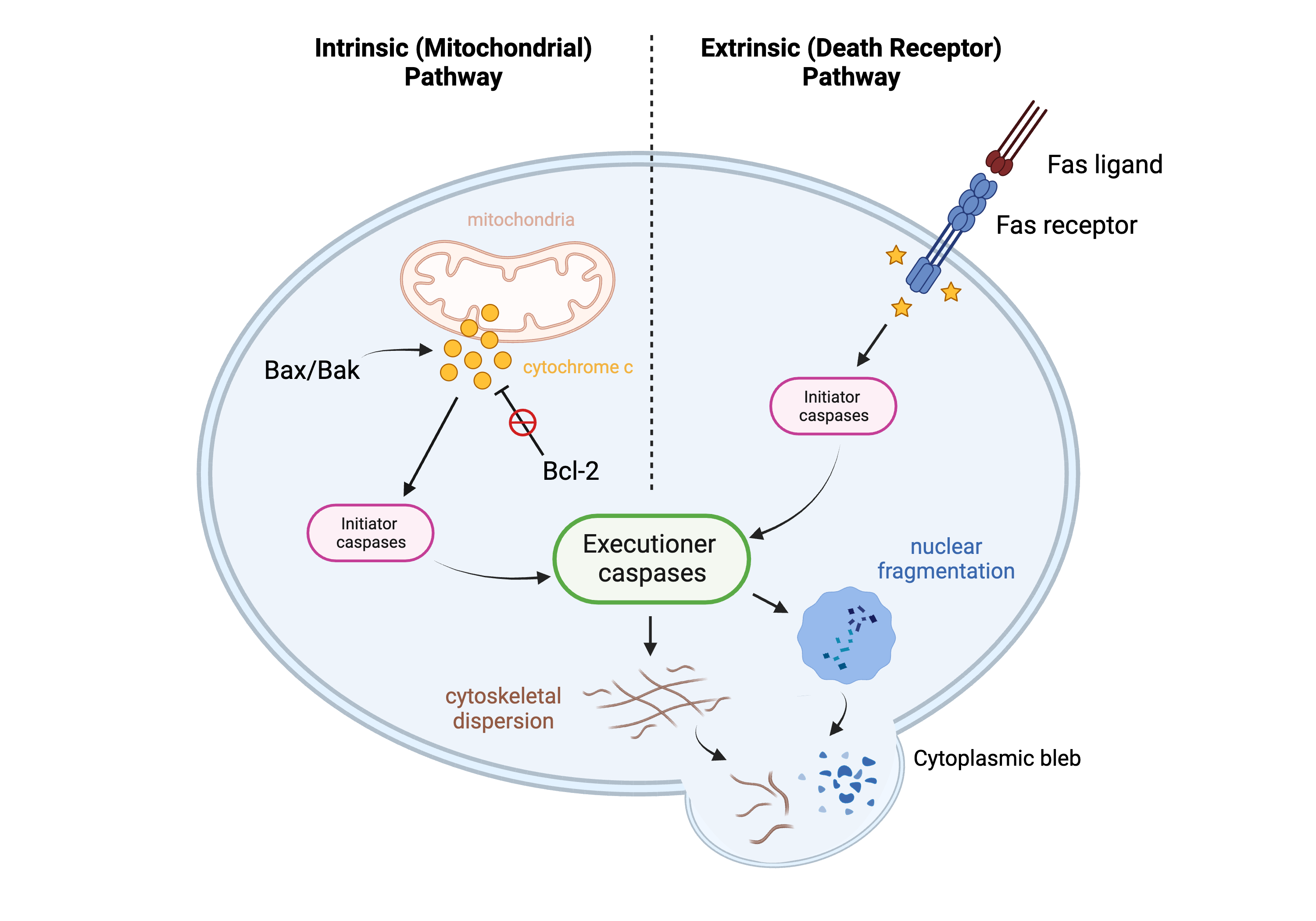
Figure 3. Apoptosis pathways. This figure demonstrates two pathways of apoptosis: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway. Created in BioRender.com.
A defining feature of apoptosis is that the programmed cell death event does not trigger inflammation: the process occurs neatly and “quietly.” This highly coordinated pathway involves two main groups of players that work with each other in concert to ensure that this organized process remains balanced: apoptosis activators and apoptosis regulators. There are two main pathways of apoptosis activation: the extrinsic pathway and the intrinsic pathway [62]. Both pathways involve an apoptotic signal, which activates a proteinase called an initiator caspase, which in turn activates another proteinase called an executioner caspase [62]. The two pathways differ in how the initiator caspase becomes activated, but the end result remains the same. The extrinsic pathway, also known as the death receptor pathway, involves the Fas ligand (FasL) binding to a cell’s Fas receptor; the intrinsic pathway, also known as the mitochondrial pathway, involves the mitochondrial release of cytochrome c into the cytoplasm, which carries out effector functions necessary for apoptosis completion [63]. Two proteins that assist in the activation of apoptosis (and hence are considered “pro-apoptotic” proteins) are Bax and Bak, and their interaction with each other triggers destabilization of the mitochondrial membrane (via oligomerization) and the subsequent release of cytochrome c into the cytoplasm [62][64]. If Bax and Bak do not interact/oligomerize, the mitochondrial membrane remains stabilized, and there is no release of cytochrome c; if Bax and Bak do oligomerize, the membrane becomes destabilized, leading to the release of cytochrome c into the cytoplasm and completion of apoptosis [64]. Equally as important as apoptosis activation is the regulation of apoptosis by proteins within the Bcl-2 family. Bcl-2 is a regulatory protein within this family that functions as a direct regulator of Bax and Bak. Bcl-2 sequesters Bax and Bak so that they cannot oligomerize; this function earned Bcl-2 its “anti-apoptotic” title [63]. The level of Bcl-2, therefore, is a good biomarker to study changes in apoptosis within cells.
While some or all of the processes may be occurring in some way during the sepsis spectrum, they are difficult to definitively test for in a clinical setting. It is clear that cell death mechanisms are occurring in sepsis patients, as one of the main features of late sepsis is a decreased white blood cell count, and more specifically, lymphopenia [65].
While lymphocyte apoptosis is a normal physiologic process necessary for proper immune functioning (i.e., a crucial part of the central tolerance of lymphocytes), this process can become dysregulated or can be hijacked by microbes, leading to an unnecessary loss of lymphocytes. Microbes can induce apoptosis in lymphocytes and other types of leukocytes in order to artificially create a more permissive environment for their survival within the host cells [66]. By triggering apoptosis within lymphocytes, microbes are able to thrive in the body because there are fewer immune cells capable of clearing them from the host. For example, Bacillus anthracis, the causative agent of anthrax, releases lethal toxin (LTx), which can lead to direct caspase release in macrophages, deceptively triggering their cell death pathways [67]. In the CLP model of polymicrobial sepsis, there is ~10% increase in the number of apoptotic lymphocytes compared with control mice [66]. Studies have shown that increased expression of Bcl-2 leads to increased survival of CLP-septic mice by ~40–50%, specifically through apoptosis prevention in splenic lymphocytes [66][68][69]. Additionally, there are numerous accounts confirming apoptosis of lymphocytes in CLP-septic mice using specific transferase-mediated dUTP nick-end labeling (TUNEL) staining methods [70][71][72].
Another mechanism of lymphocyte suppression that falls outside of, but is closely related to, the apoptotic pathways is the PD-1/PD-L1 pathway that was described earlier. While this pathway does not lead to apoptosis of lymphocytes, it renders the lymphocytes inactive, producing a very similar outcome. While the application of checkpoint inhibitors (i.e., PD-1 inhibitors) for sepsis-induced immunosuppression is still in its nascent stages, the enhancement of lymphocyte functionality in late sepsis may provide an exciting avenue for researchers moving forward.
In an attempt to target sepsis-induced immunosuppression, there have been several clinical trials whereby lymphocytes were targeted for restoration. In one study, recombinant GM-CSF or G-CSF was administered to late-septic patients in order to increase their lymphocyte counts [73]. These attempts initially seemed promising, but the beneficial effects did not persist long-term. In a similar clinical trial, IL-7 was administered to late-septic patients in an attempt to increase their populations of T lymphocytes, but the patients did not improve long-term [74]. In both of these instances, the failures were attributed to pathological migration and activation of myeloid-derived suppressor cells (MDSCs) [75]. It was postulated that MDSCs suppressed T lymphocyte activity and proliferation, negating the beneficial action of the intervention.
3. Myeloid-Derived Suppressor Cells
The third hallmark of sepsis-induced immunosuppression is increased myeloid-derived suppressor cell (MDSC) production/migration [1]. MDSCs are a group of heterogenous, immature cells of the myeloid lineage that work to suppress the innate and adaptive immune systems [76]. MDSCs can be divided into two main populations based on their lineage (although there are others that have been reported [77]): monocytic MDSCs (M-MDSCs) and polymorphic MDSCs (PMN-MDSCs) [76][78][79]. Both M-MDSCs and PMN-MDSCs are able to exert their inhibitory effects on the innate and adaptive immune systems in similar ways, with the primary mechanisms being via the release of ROS, production of arginase-1, and release of the anti-inflammatory cytokine IL-10 [76]. MDSCs exist in the bone marrow and are triggered to migrate into the blood and secondary lymphoid tissues via chemokine CXCL2 and IL-8 gradients [76]. Overall, MDSCs function as immunosuppressive cells, but their exact role and the meaning behind their migration patterns in patients with sepsis remains to be seen.
There is some controversy regarding the significance of the MDSC migration that is observed in septic patients [80][81][82][83]. Some studies report that the increase in MDSC migration out of the bone marrow in sepsis patients is partly responsible for the adverse clinical outcomes due to late sepsis [75][83][84][85]. In fact, Tang et al. suggests that inhibiting MDSCs would be one of the most effective treatments for sepsis-induced immunosuppression overall [85]. In septic patients, having elevated numbers of MDSCs in the peripheral blood was associated in some patients with longer ICU hospital stays and a higher statistical likelihood of acquiring a secondary infection [75]. In LPS-induced immunosuppression, MDSCs migrate from the bone marrow to the blood/secondary lymphoid tissues and inhibit the proliferation of lymphocytes [84]. Additional studies have shown that not only are MDSCs directly inhibitory on lymphocytes, but also their production of arginase-1 further disrupts T-cell functions [86]. Similarly, MDSCs can indirectly cause immunosuppression by activating Tregs [75]. This anti-inflammatory T-cell subset functions to inhibit other innate and adaptive immune cells, primarily via the release of IL-10 [75].
Conversely, there are also reports that MDSC migration during late sepsis could actually contribute to long-term survival in sepsis patients [80][81][83][87][88]. This paradoxical function of MDSCs was studied extensively in CLP-sepsis models. In one study, MDSCs were adoptively transferred into CLP mice early on in the infection timeline, and these mice had decreased mortality compared with sham animals [80]. In another study, mouse MDSC expansion was inhibited by pretreatment with the chemotherapeutic gemcitabine, and survival to CLP was dramatically reduced in the gemcitabine treatment group compared with the sham animals [81]. Similarly, in another study, inhibition of CLP mouse MDSC populations via GR-1 neutralization led to decreased survival compared with the sham animals [88]. In another infection model system, in vivo depletion of MDSCs in mice infected with Trypanosoma cruzi (the causative agent of Chagas disease) led to increased mortality compared with the sham animals [87].
A study by Schrijver et al. provides a strong case for the expansion and migration of M-MDSCs in particular correlating with improved survival in sepsis patients [89]. Blood was taken from patients in eight ICUs with pneumonia secondary to sepsis [89]. The authors found that patients with high levels of M-MDSCs overall had significantly reduced 90-day mortality rates and improved survival compared with patients with high levels of PMN-MDSCs [89]. The contention is that due to the heterogeneity of MDSC populations, perhaps overall, MDSCs play a dual role during sepsis, with each unique MDSC population evolving over time after sepsis onset [80][89][90].
Overall, in sepsis patients, there is an elevation in MDSC numbers in the blood and the secondary lymphoid tissues. While a simple explanation would be that the migrating MDSCs are acting as the trigger for sepsis-induced immunosuppression, perhaps what is actually occurring here is that the host is attempting to correct the early hyper-proinflammatory phase of sepsis with MDSC migration. Due to unidentified dysregulation pathways in sepsis patients, perhaps the recruitment of MDSCs is not robust enough in these particular patients; therefore, the hyper-proinflammatory response persists until the late stage of sepsis sets in, and their presence then accentuates the immunosuppression. MDSC expansion and migration in sepsis appears to be a highly dynamic process, and these cells, conceivably, are malleable to the infection at hand. Although the complete consequences of increased MDSC production in sepsis have not been fully elucidated, their presence in the blood as a biomarker in the immunosuppressive phase of sepsis cannot be disputed.
References
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-Induced Immunosuppression: Mechanisms, Diagnosis and Current Treatment Options. Mil. Med. Res. 2022, 9, 56.
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1.
- Lam, R.S.; O’Brien-Simpson, N.M.; Holden, J.A.; Lenzo, J.C.; Fong, S.B.; Reynolds, E.C. Unprimed, M1 and M2 Macrophages Differentially Interact with Porphyromonas Gingivalis. PLoS ONE 2016, 11, e0158629.
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460.
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 Macrophages and Their Overlaps—Myth or Reality? Clin. Sci. 2023, 137, 1067–1093.
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The Role of Macrophages in the Resolution of Inflammation. J. Clin. Investig. 2019, 129, 2619–2628.
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795.
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792.
- Zhang, Q.; Sioud, M. Tumor-Associated Macrophage Subsets: Shaping Polarization and Targeting. Int. J. Mol. Sci. 2023, 24, 7493.
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613.
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417.
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288.
- Saas, P.; Chagué, C.; Maraux, M.; Cherrier, T. Toward the Characterization of Human Pro-Resolving Macrophages? Front. Immunol. 2020, 11, 593300.
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617.
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483.
- Cronan, M.R.; Beerman, R.W.; Rosenberg, A.F.; Saelens, J.W.; Johnson, M.G.; Oehlers, S.H.; Sisk, D.M.; Jurcic Smith, K.L.; Medvitz, N.A.; Miller, S.E.; et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity 2016, 45, 861–876.
- Linke, M.; Pham, H.T.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic Signaling via the Metabolic Checkpoint Kinase mTORC1 Induces Macrophage Granuloma Formation and Marks Sarcoidosis Progression. Nat. Immunol. 2017, 18, 293–302.
- Zhang, T.; Yu-Jing, L.; Ma, T. Role of Regulation of PD-1 and PD-L1 Expression in Sepsis. Front. Immunol. 2023, 14, 1029438.
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10.
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561.
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 Co-stimulation Contributes to Ligand-induced T Cell Receptor Down-modulation on CD8 + T Cells. EMBO Mol. Med. 2011, 3, 581–592.
- Song, S.; Yuan, P.; Wu, H.; Chen, J.; Fu, J.; Li, P.; Lu, J.; Wei, W. Dendritic Cells with an Increased PD-L1 by TGF-β Induce T Cell Anergy for the Cytotoxicity of Hepatocellular Carcinoma Cells. Int. Immunopharmacol. 2014, 20, 117–123.
- Zhou, X.; Zhou, Y.; Ding, Q.; Jiao, Z.; Lu, L.; Yang, N.; Ma, Y.; Chou, K.-Y. High Level Expression of B7H1 Molecules by Keratinocytes Suppresses Xeno- and Allo-Reactions by Inducing Type I Regulatory T Cells. Transplant. Immunol. 2009, 21, 192–197.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Crouser, E.D.; Martin, G.S.; Albertson, T.; Bartz, R.R.; Brakenridge, S.C.; Delano, M.J.; Park, P.K.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Nivolumab. Intensive Care Med. 2019, 45, 1360–1371.
- Yang, L.; Gao, Q.; Li, Q.; Guo, S. PD-L1 Blockade Improves Survival in Sepsis by Reversing Monocyte Dysfunction and Immune Disorder. Inflammation 2023, 1–15.
- Ruan, W.-S.; Feng, M.-X.; Xu, J.; Xu, Y.-G.; Song, C.-Y.; Lin, L.-Y.; Li, L.; Lu, Y.-Q. Early Activation of Myeloid-Derived Suppressor Cells Participate in Sepsis-Induced Immune Suppression via PD-L1/PD-1 Axis. Front. Immunol. 2020, 11, 1299.
- Patera, A.C.; Drewry, A.M.; Chang, K.; Beiter, E.R.; Osborne, D.; Hotchkiss, R.S. Frontline Science: Defects in Immune Function in Patients with Sepsis Are Associated with PD-1 or PD-L1 Expression and Can Be Restored by Antibodies Targeting PD-1 or PD-L1. J. Leukoc. Biol. 2016, 100, 1239–1254.
- Wilson, J.K.; Zhao, Y.; Singer, M.; Spencer, J.; Shankar-Hari, M. Lymphocyte Subset Expression and Serum Concentrations of PD-1/PD-L1 in Sepsis—Pilot Study. Crit. Care 2018, 22, 95.
- Zhang, Y.; Li, J.; Lou, J.; Zhou, Y.; Bo, L.; Zhu, J.; Zhu, K.; Wan, X.; Cai, Z.; Deng, X. Upregulation of Programmed Death-1 on T Cells and Programmed Death Ligand-1 on Monocytes in Septic Shock Patients. Crit. Care 2011, 15, R70.
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin Tolerance: New Mechanisms, Molecules and Clinical Significance. Trends Immunol. 2009, 30, 475–487.
- Cavaillon, J.-M.; Adib-Conquy, M. Bench-to-Bedside Review: Endotoxin Tolerance as a Model of Leukocyte Reprogramming in Sepsis. Crit. Care 2006, 10, 233.
- Hoppstädter, J.; Dembek, A.; Linnenberger, R.; Dahlem, C.; Barghash, A.; Fecher-Trost, C.; Fuhrmann, G.; Koch, M.; Kraegeloh, A.; Huwer, H.; et al. Toll-Like Receptor 2 Release by Macrophages: An Anti-Inflammatory Program Induced by Glucocorticoids and Lipopolysaccharide. Front. Immunol. 2019, 10, 1634.
- Pradhan, K.; Yi, Z.; Geng, S.; Li, L. Development of Exhausted Memory Monocytes and Underlying Mechanisms. Front. Immunol. 2021, 12, 778830.
- Cavaillon, J.M.; Adib-Conquy, M.; Cloëz-Tayarani, I.; Fitting, C. Immunodepression in Sepsis and SIRS Assessed by Ex Vivo Cytokine Production Is Not a Generalized Phenomenon: A Review. J. Endotoxin Res. 2001, 7, 85–93.
- Bick, A.; Buys, W.; Engler, A.; Madel, R.; Atia, M.; Faro, F.; Westendorf, A.M.; Limmer, A.; Buer, J.; Herbstreit, F.; et al. Immune Hyporeactivity to Bacteria and Multiple TLR-Ligands, yet No Response to Checkpoint Inhibition in Patients Just after Meeting Sepsis-3 Criteria. PLoS ONE 2022, 17, e0273247.
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA 2011, 306, 2594.
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874.
- Blackwell, T.S.; Blackwell, T.R.; Christman, J.W. Induction of Endotoxin Tolerance Depletes Nuclear Factor-κB and Suppresses Its Activation in Rat Alveolar Macrophages. J. Leukoc. Biol. 1997, 62, 885–891.
- Chan, C.; Li, L.; McCall, C.E.; Yoza, B.K. Endotoxin Tolerance Disrupts Chromatin Remodeling and NF-κB Transactivation at the IL-1β Promoter. J. Immunol. 2005, 175, 461–468.
- Del Fresno, C.; García-Rio, F.; Gómez-Piña, V.; Soares-Schanoski, A.; Fernández-Ruíz, I.; Jurado, T.; Kajiji, T.; Shu, C.; Marín, E.; Gutierrez Del Arroyo, A.; et al. Potent Phagocytic Activity with Impaired Antigen Presentation Identifying Lipopolysaccharide-Tolerant Human Monocytes: Demonstration in Isolated Monocytes from Cystic Fibrosis Patients. J. Immunol. 2009, 182, 6494–6507.
- Deng, H.; Maitra, U.; Morris, M.; Li, L. Molecular Mechanism Responsible for the Priming of Macrophage Activation. J. Biol. Chem. 2013, 288, 3897–3906.
- Dobrovolskaia, M.A.; Vogel, S.N. Toll Receptors, CD14, and Macrophage Activation and Deactivation by LPS. Microbes Infect. 2002, 4, 903–914.
- Foster, S.L.; Medzhitov, R. Gene-Specific Control of the TLR-Induced Inflammatory Response. Clin. Immunol. 2009, 130, 7–15.
- Hoogendijk, A.J.; Garcia-Laorden, M.I.; Van Vught, L.A.; Wiewel, M.A.; Belkasim-Bohoudi, H.; Duitman, J.; Horn, J.; Schultz, M.J.; Scicluna, B.P.; Van ‘T Veer, C.; et al. Sepsis Patients Display a Reduced Capacity to Activate Nuclear Factor-κB in Multiple Cell Types*. Crit. Care Med. 2017, 45, e524–e531.
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.-S.; et al. PD-1 Expression by Macrophages Plays a Pathologic Role in Altering Microbial Clearance and the Innate Inflammatory Response to Sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308.
- Zhou, M.; Aziz, M.; Yen, H.-T.; Ma, G.; Murao, A.; Wang, P. Extracellular CIRP Dysregulates Macrophage Bacterial Phagocytosis in Sepsis. Cell Mol. Immunol. 2022, 20, 80–93.
- Chen, W.; Qiang, X.; Wang, Y.; Zhu, S.; Li, J.; Babaev, A.; Yang, H.; Gong, J.; Becker, L.; Wang, P.; et al. Identification of Tetranectin-Targeting Monoclonal Antibodies to Treat Potentially Lethal Sepsis. Sci. Transl. Med. 2020, 12, eaaz3833.
- Yang, G.; Zhang, Q.; Tan, J.; Xiong, Y.; Liang, Y.; Yan, J.; Gu, F.; Xu, Y. HMGB1 Induces Macrophage Pyroptosis in Chronic Endometritis. Int. Immunopharmacol. 2023, 123, 110706.
- Zhang, W.; Jiang, H.; Wu, G.; Huang, P.; Wang, H.; An, H.; Liu, S.; Zhang, W. The Pathogenesis and Potential Therapeutic Targets in Sepsis. MedComm 2023, 4, e418.
- Wang, Y.; Zhang, W.; Xu, Y.; Wu, D.; Gao, Z.; Zhou, J.; Qian, H.; He, B.; Wang, G. Extracellular HMGB1 Impairs Macrophage-Mediated Efferocytosis by Suppressing the Rab43-Controlled Cell Surface Transport of CD91. Front. Immunol. 2022, 13, 767630.
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 Enhances Immune Suppression by Facilitating the Differentiation and Suppressive Activity of Myeloid-Derived Suppressor Cells. Cancer Res. 2014, 74, 5723–5733.
- Döcke, W.-D.; Randow, F.; Syrbe, U.; Krausch, D.; Asadullah, K.; Reinke, P.; Volk, H.-D.; Kox, W. Monocyte Deactivation in Septic Patients: Restoration by IFN-γ Treatment. Nat. Med. 1997, 3, 678–681.
- Kim, E.Y.; Ner-Gaon, H.; Varon, J.; Cullen, A.M.; Guo, J.; Choi, J.; Barragan-Bradford, D.; Higuera, A.; Pinilla-Vera, M.; Short, S.A.P.; et al. Post-Sepsis Immunosuppression Depends on NKT Cell Regulation of mTOR/IFN-γ in NK Cells. J. Clin. Investig. 2020, 130, 3238–3252.
- Shen, S.; Shao, Y.; Li, C. Different Types of Cell Death and Their Shift in Shaping Disease. Cell Death Discov. 2023, 9, 284.
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, Ferroptosis, Pyroptosis, and Necroptosis in Tumor Immunotherapy. Sig Transduct. Target. Ther. 2022, 7, 196.
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell Mol. Immunol. 2021, 18, 1106–1121.
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364.
- Rathmell, J.C.; Thompson, C.B. Pathways of Apoptosis in Lymphocyte Development, Homeostasis, and Disease. Cell 2002, 109, S97–S107.
- Alberts, B. (Ed.) Molecular Biology of the Cell, 4th ed.; Garland: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3.
- Tsujimoto, Y. Role of Bcl-2 Family Proteins in Apoptosis: Apoptosomes or Mitochondria?: Role of Bcl-2 Family Proteins in Apoptosis. Genes. Cells 1998, 3, 697–707.
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431.
- Sheikh Motahar Vahedi, H.; Bagheri, A.; Jahanshir, A.; Seyedhosseini, J.; Vahidi, E. Association of Lymphopenia with Short Term Outcomes of Sepsis Patients; a Brief Report. Arch. Acad. Emerg. Med. 2019, 7, e14.
- Carrero, J.; Unanue, E. Lymphocyte Apoptosis as an Immune Subversion Strategy of Microbial Pathogens. Trends Immunol. 2006, 27, 497–503.
- Van Hauwermeiren, F.; Van Opdenbosch, N.; Van Gorp, H.; De Vasconcelos, N.; Van Loo, G.; Vandenabeele, P.; Kanneganti, T.-D.; Lamkanfi, M. Bacillus Anthracis Induces NLRP3 Inflammasome Activation and Caspase-8–Mediated Apoptosis of Macrophages to Promote Lethal Anthrax. Proc. Natl. Acad. Sci. USA 2022, 119, e2116415119.
- Hotchkiss, R.S.; Swanson, P.E.; Knudson, C.M.; Chang, K.C.; Cobb, J.P.; Osborne, D.F.; Zollner, K.M.; Buchman, T.G.; Korsmeyer, S.J.; Karl, I.E. Overexpression of Bcl-2 in Transgenic Mice Decreases Apoptosis and Improves Survival in Sepsis. J. Immunol. 1999, 162, 4148–4156.
- Wang, S.D.; Huang, K.J.; Lin, Y.S.; Lei, H.Y. Sepsis-Induced Apoptosis of the Thymocytes in Mice. J. Immunol. 1994, 152, 5014–5021.
- Luan, Y.; Yao, Y.; Xiao, X.; Sheng, Z. Insights into the Apoptotic Death of Immune Cells in Sepsis. J. Interferon Cytokine Res. 2015, 35, 17–22.
- Luan, Y.-Y.; Dong, N.; Xie, M.; Xiao, X.-Z.; Yao, Y.-M. The Significance and Regulatory Mechanisms of Innate Immune Cells in the Development of Sepsis. J. Interferon Cytokine Res. 2014, 34, 2–15.
- Matsuda, N.; Teramae, H.; Futatsugi, M.; Takano, K.; Yamamoto, S.; Tomita, K.; Suzuki, T.; Yokoo, H.; Koike, K.; Hattori, Y. Up-Regulation of Histamine H 4 Receptors Contributes to Splenic Apoptosis in Septic Mice: Counteraction of the Antiapoptotic Action of Nuclear Factor-κB. J. Pharmacol. Exp. Ther. 2010, 332, 730–737.
- Bo, L.; Wang, F.; Zhu, J.; Li, J.; Deng, X. Granulocyte-Colony Stimulating Factor (G-CSF) and Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) for Sepsis: A Meta-Analysis. Crit. Care 2011, 15, R58.
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmelé, T.; Blood, T.; Morre, M.; et al. Interleukin-7 Restores Lymphocytes in Septic Shock: The IRIS-7 Randomized Clinical Trial. JCI Insight 2018, 3, e98960.
- Zhang, W.; Fang, X.; Gao, C.; Song, C.; He, Y.; Zhou, T.; Yang, X.; Shang, Y.; Xu, J. MDSCs in Sepsis-Induced Immunosuppression and Its Potential Therapeutic Targets. Cytokine Growth Factor. Rev. 2023, 69, 90–103.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Goldmann, O.; Beineke, A.; Medina, E. Identification of a Novel Subset of Myeloid-Derived Suppressor Cells During Chronic Staphylococcal Infection That Resembles Immature Eosinophils. J. Infect. Dis. 2017, 216, 1444–1451.
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150.
- Talmadge, J.E.; Gabrilovich, D.I. History of Myeloid-Derived Suppressor Cells. Nat. Rev. Cancer 2013, 13, 739–752.
- Brudecki, L.; Ferguson, D.A.; McCall, C.E.; El Gazzar, M. Myeloid-Derived Suppressor Cells Evolve during Sepsis and Can Enhance or Attenuate the Systemic Inflammatory Response. Infect. Immun. 2012, 80, 2026–2034.
- Cuenca, A.G.; Delano, M.J.; Kelly-Scumpia, K.M.; Moreno, C.; Scumpia, P.O.; LaFace, D.M.; Heyworth, P.G.; Efron, P.A.; Moldawer, L.L. A Paradoxical Role for Myeloid-Derived Suppressor Cells in Sepsis and Trauma. Mol. Med. 2011, 17, 281–292.
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.L.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-Dependent Expansion of an Immature GR-1+CD11b+ Population Induces T Cell Suppression and Th2 Polarization in Sepsis. J. Exp. Med. 2007, 204, 1463–1474.
- Mathias, B.; Delmas, A.L.; Ozrazgat-Baslanti, T.; Vanzant, E.L.; Szpila, B.E.; Mohr, A.M.; Moore, F.A.; Brakenridge, S.C.; Brumback, B.A.; Moldawer, L.L.; et al. Human Myeloid-Derived Suppressor Cells Are Associated with Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann. Surg. 2017, 265, 827–834.
- Landoni, V.I.; Martire-Greco, D.; Rodriguez-Rodrigues, N.; Chiarella, P.; Schierloh, P.; Isturiz, M.A.; Fernández, G.C. Immature Myeloid Gr-1+ CD11b+ Cells from Lipopolysaccharide-Immunosuppressed Mice Acquire Inhibitory Activity in the Bone Marrow and Migrate to Lymph Nodes to Exert Their Suppressive Function. Clin. Sci. 2016, 130, 259–271.
- Tang, H.; Li, H.; Sun, Z. Targeting Myeloid-Derived Suppressor Cells for Cancer Therapy. Cancer Biol. Med. 2021, 18, 992–1009.
- Uhel, F.; Azzaoui, I.; Grégoire, M.; Pangault, C.; Dulong, J.; Tadié, J.-M.; Gacouin, A.; Camus, C.; Cynober, L.; Fest, T.; et al. Early Expansion of Circulating Granulocytic Myeloid-Derived Suppressor Cells Predicts Development of Nosocomial Infections in Patients with Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 315–327.
- Arocena, A.R.; Onofrio, L.I.; Pellegrini, A.V.; Carrera Silva, A.E.; Paroli, A.; Cano, R.C.; Aoki, M.P.; Gea, S. Myeloid-derived Suppressor Cells Are Key Players in the Resolution of Inflammation during a Model of Acute Infection. Eur. J. Immunol. 2014, 44, 184–194.
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic Acute-Phase Proteins Control Innate Immune Responses during Infection by Promoting Myeloid-Derived Suppressor Cell Function. J. Exp. Med. 2010, 207, 1453–1464.
- Schrijver, I.T.; Karakike, E.; Théroude, C.; Baumgartner, P.; Harari, A.; Giamarellos-Bourboulis, E.J.; Calandra, T.; Roger, T. High Levels of Monocytic Myeloid-Derived Suppressor Cells Are Associated with Favorable Outcome in Patients with Pneumonia and Sepsis with Multi-Organ Failure. Intensive Care Med. Exp. 2022, 10, 5.
- Hollen, M.K.; Stortz, J.A.; Darden, D.; Dirain, M.L.; Nacionales, D.C.; Hawkins, R.B.; Cox, M.C.; Lopez, M.-C.; Rincon, J.C.; Ungaro, R.; et al. Myeloid-Derived Suppressor Cell Function and Epigenetic Expression Evolves over Time after Surgical Sepsis. Crit. Care 2019, 23, 355.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
807
Revisions:
2 times
(View History)
Update Date:
01 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No