Severe infection can lead to sepsis. In sepsis, the host mounts an inappropriately large inflammatory response in an attempt to clear the invading pathogen. This sustained high level of inflammation may cause tissue injury and organ failure. Later in sepsis, a paradoxical immunosuppression occurs, where the host is unable to clear the preexisting infection and is susceptible to secondary infections. A major issue with sepsis treatment is that it is difficult for physicians to ascertain which stage of sepsis the patient is in. Sepsis treatment will depend on the patient’s immune status across the spectrum of the disease, and these immune statuses are nearly polar opposites in the early and late stages of sepsis.
- macrophages
- lymphocytes
- apoptosis
- infection
- MDSCs
1. Monocyte/Macrophage Exhaustion
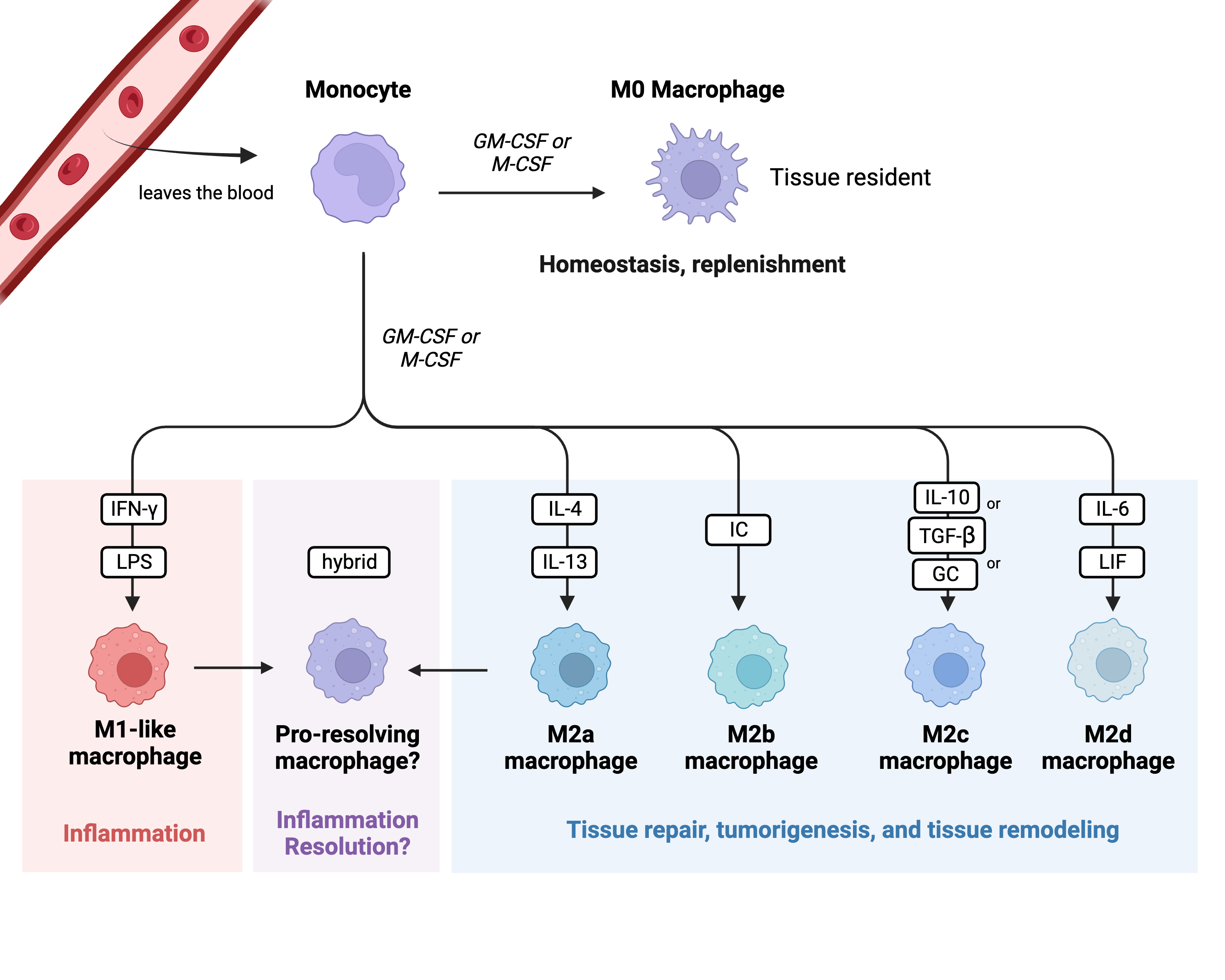
1.1. PD-1 and PDL-1
1.2. Exhausted Monocytes/Macrophages
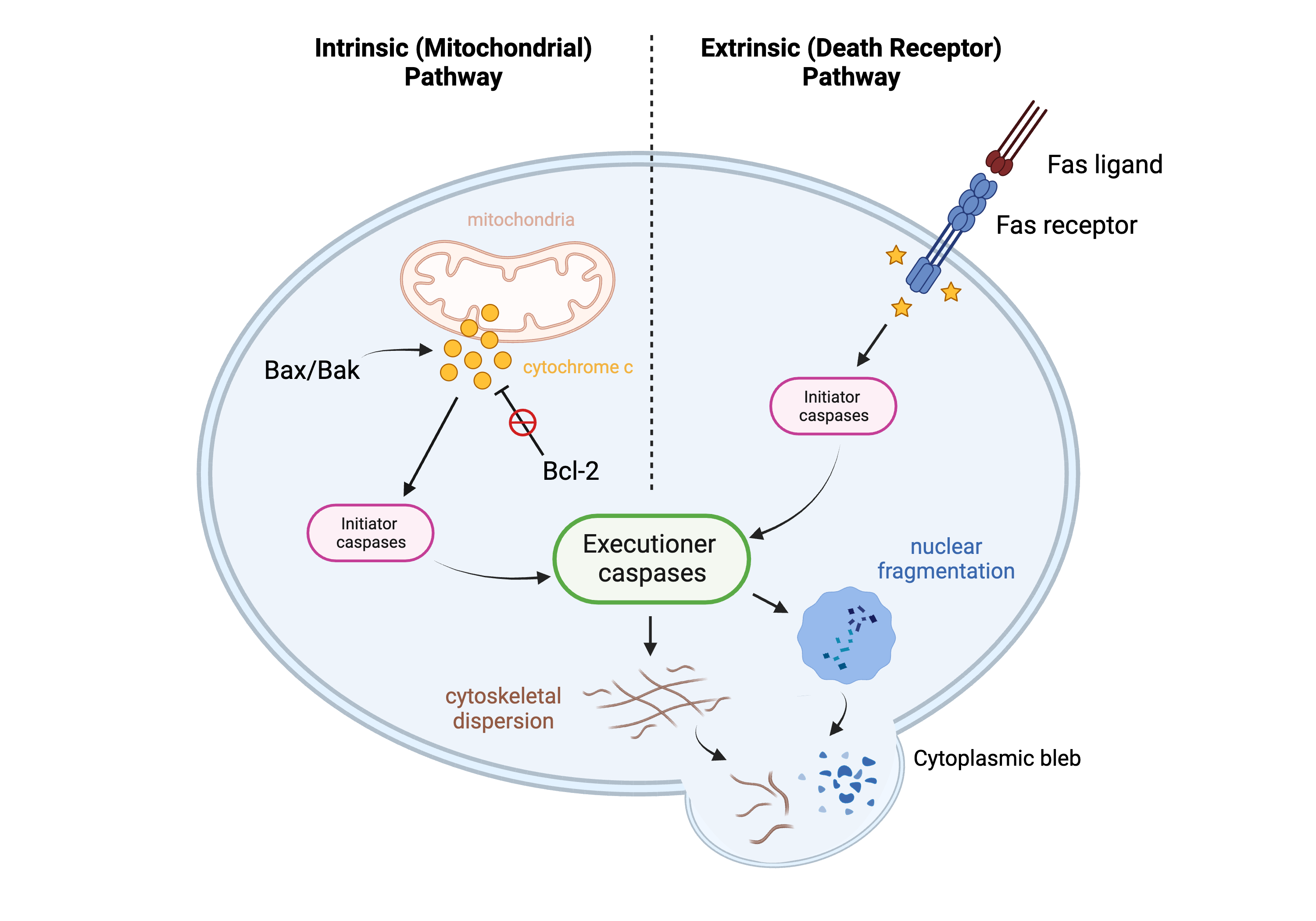
2. Lymphocyte Apoptosis
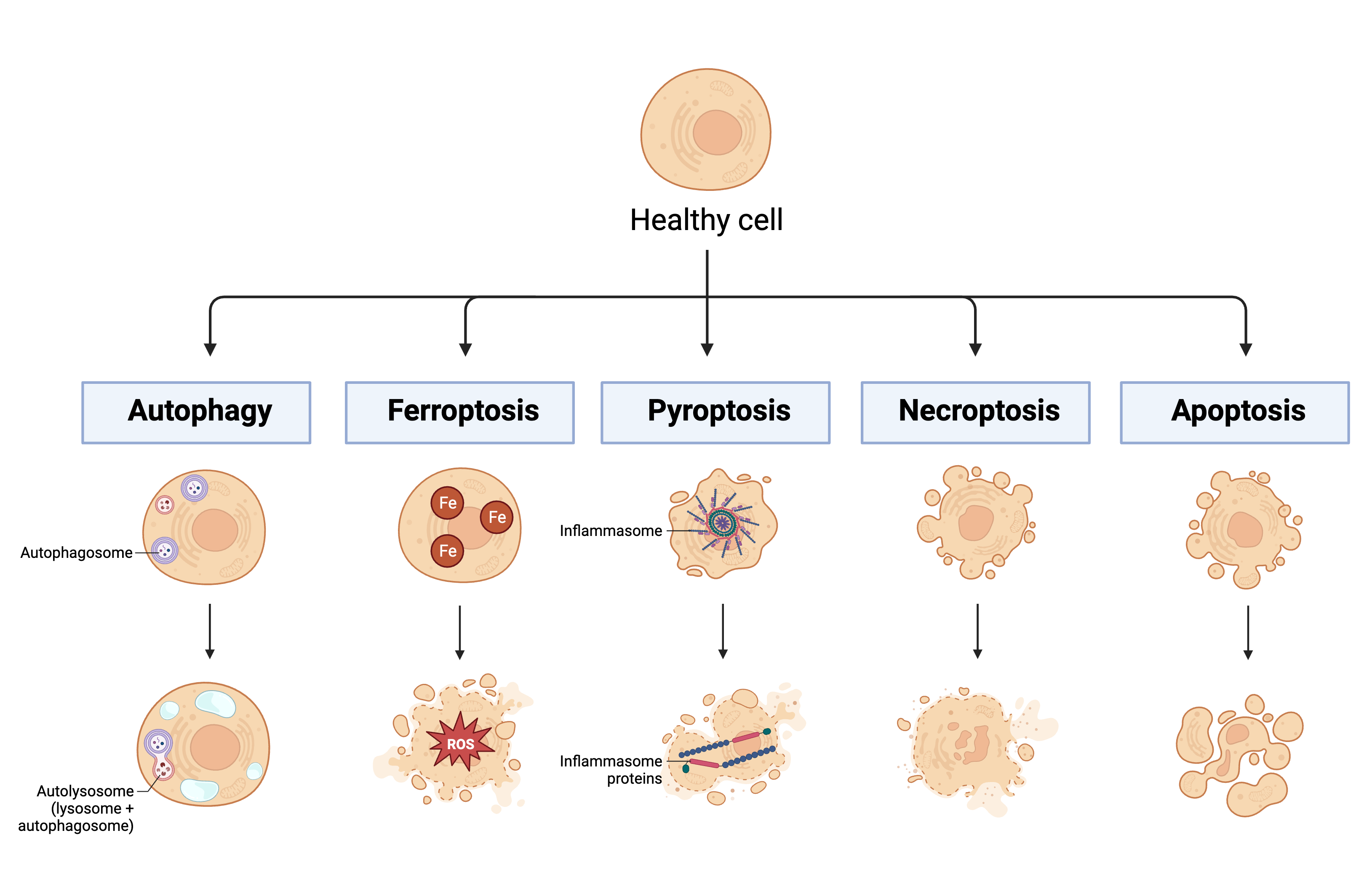
3. Myeloid-Derived Suppressor Cells
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines12010175
References
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-Induced Immunosuppression: Mechanisms, Diagnosis and Current Treatment Options. Mil. Med. Res. 2022, 9, 56.
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1.
- Lam, R.S.; O’Brien-Simpson, N.M.; Holden, J.A.; Lenzo, J.C.; Fong, S.B.; Reynolds, E.C. Unprimed, M1 and M2 Macrophages Differentially Interact with Porphyromonas Gingivalis. PLoS ONE 2016, 11, e0158629.
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460.
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 Macrophages and Their Overlaps—Myth or Reality? Clin. Sci. 2023, 137, 1067–1093.
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The Role of Macrophages in the Resolution of Inflammation. J. Clin. Investig. 2019, 129, 2619–2628.
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795.
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792.
- Zhang, Q.; Sioud, M. Tumor-Associated Macrophage Subsets: Shaping Polarization and Targeting. Int. J. Mol. Sci. 2023, 24, 7493.
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613.
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417.
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288.
- Saas, P.; Chagué, C.; Maraux, M.; Cherrier, T. Toward the Characterization of Human Pro-Resolving Macrophages? Front. Immunol. 2020, 11, 593300.
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617.
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483.
- Cronan, M.R.; Beerman, R.W.; Rosenberg, A.F.; Saelens, J.W.; Johnson, M.G.; Oehlers, S.H.; Sisk, D.M.; Jurcic Smith, K.L.; Medvitz, N.A.; Miller, S.E.; et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity 2016, 45, 861–876.
- Linke, M.; Pham, H.T.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic Signaling via the Metabolic Checkpoint Kinase mTORC1 Induces Macrophage Granuloma Formation and Marks Sarcoidosis Progression. Nat. Immunol. 2017, 18, 293–302.
- Zhang, T.; Yu-Jing, L.; Ma, T. Role of Regulation of PD-1 and PD-L1 Expression in Sepsis. Front. Immunol. 2023, 14, 1029438.
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10.
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561.
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 Co-stimulation Contributes to Ligand-induced T Cell Receptor Down-modulation on CD8 + T Cells. EMBO Mol. Med. 2011, 3, 581–592.
- Song, S.; Yuan, P.; Wu, H.; Chen, J.; Fu, J.; Li, P.; Lu, J.; Wei, W. Dendritic Cells with an Increased PD-L1 by TGF-β Induce T Cell Anergy for the Cytotoxicity of Hepatocellular Carcinoma Cells. Int. Immunopharmacol. 2014, 20, 117–123.
- Zhou, X.; Zhou, Y.; Ding, Q.; Jiao, Z.; Lu, L.; Yang, N.; Ma, Y.; Chou, K.-Y. High Level Expression of B7H1 Molecules by Keratinocytes Suppresses Xeno- and Allo-Reactions by Inducing Type I Regulatory T Cells. Transplant. Immunol. 2009, 21, 192–197.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Crouser, E.D.; Martin, G.S.; Albertson, T.; Bartz, R.R.; Brakenridge, S.C.; Delano, M.J.; Park, P.K.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Nivolumab. Intensive Care Med. 2019, 45, 1360–1371.
- Yang, L.; Gao, Q.; Li, Q.; Guo, S. PD-L1 Blockade Improves Survival in Sepsis by Reversing Monocyte Dysfunction and Immune Disorder. Inflammation 2023, 1–15.
- Ruan, W.-S.; Feng, M.-X.; Xu, J.; Xu, Y.-G.; Song, C.-Y.; Lin, L.-Y.; Li, L.; Lu, Y.-Q. Early Activation of Myeloid-Derived Suppressor Cells Participate in Sepsis-Induced Immune Suppression via PD-L1/PD-1 Axis. Front. Immunol. 2020, 11, 1299.
- Patera, A.C.; Drewry, A.M.; Chang, K.; Beiter, E.R.; Osborne, D.; Hotchkiss, R.S. Frontline Science: Defects in Immune Function in Patients with Sepsis Are Associated with PD-1 or PD-L1 Expression and Can Be Restored by Antibodies Targeting PD-1 or PD-L1. J. Leukoc. Biol. 2016, 100, 1239–1254.
- Wilson, J.K.; Zhao, Y.; Singer, M.; Spencer, J.; Shankar-Hari, M. Lymphocyte Subset Expression and Serum Concentrations of PD-1/PD-L1 in Sepsis—Pilot Study. Crit. Care 2018, 22, 95.
- Zhang, Y.; Li, J.; Lou, J.; Zhou, Y.; Bo, L.; Zhu, J.; Zhu, K.; Wan, X.; Cai, Z.; Deng, X. Upregulation of Programmed Death-1 on T Cells and Programmed Death Ligand-1 on Monocytes in Septic Shock Patients. Crit. Care 2011, 15, R70.
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin Tolerance: New Mechanisms, Molecules and Clinical Significance. Trends Immunol. 2009, 30, 475–487.
- Cavaillon, J.-M.; Adib-Conquy, M. Bench-to-Bedside Review: Endotoxin Tolerance as a Model of Leukocyte Reprogramming in Sepsis. Crit. Care 2006, 10, 233.
- Hoppstädter, J.; Dembek, A.; Linnenberger, R.; Dahlem, C.; Barghash, A.; Fecher-Trost, C.; Fuhrmann, G.; Koch, M.; Kraegeloh, A.; Huwer, H.; et al. Toll-Like Receptor 2 Release by Macrophages: An Anti-Inflammatory Program Induced by Glucocorticoids and Lipopolysaccharide. Front. Immunol. 2019, 10, 1634.
- Pradhan, K.; Yi, Z.; Geng, S.; Li, L. Development of Exhausted Memory Monocytes and Underlying Mechanisms. Front. Immunol. 2021, 12, 778830.
- Cavaillon, J.M.; Adib-Conquy, M.; Cloëz-Tayarani, I.; Fitting, C. Immunodepression in Sepsis and SIRS Assessed by Ex Vivo Cytokine Production Is Not a Generalized Phenomenon: A Review. J. Endotoxin Res. 2001, 7, 85–93.
- Bick, A.; Buys, W.; Engler, A.; Madel, R.; Atia, M.; Faro, F.; Westendorf, A.M.; Limmer, A.; Buer, J.; Herbstreit, F.; et al. Immune Hyporeactivity to Bacteria and Multiple TLR-Ligands, yet No Response to Checkpoint Inhibition in Patients Just after Meeting Sepsis-3 Criteria. PLoS ONE 2022, 17, e0273247.
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA 2011, 306, 2594.
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874.
- Blackwell, T.S.; Blackwell, T.R.; Christman, J.W. Induction of Endotoxin Tolerance Depletes Nuclear Factor-κB and Suppresses Its Activation in Rat Alveolar Macrophages. J. Leukoc. Biol. 1997, 62, 885–891.
- Chan, C.; Li, L.; McCall, C.E.; Yoza, B.K. Endotoxin Tolerance Disrupts Chromatin Remodeling and NF-κB Transactivation at the IL-1β Promoter. J. Immunol. 2005, 175, 461–468.
- Del Fresno, C.; García-Rio, F.; Gómez-Piña, V.; Soares-Schanoski, A.; Fernández-Ruíz, I.; Jurado, T.; Kajiji, T.; Shu, C.; Marín, E.; Gutierrez Del Arroyo, A.; et al. Potent Phagocytic Activity with Impaired Antigen Presentation Identifying Lipopolysaccharide-Tolerant Human Monocytes: Demonstration in Isolated Monocytes from Cystic Fibrosis Patients. J. Immunol. 2009, 182, 6494–6507.
- Deng, H.; Maitra, U.; Morris, M.; Li, L. Molecular Mechanism Responsible for the Priming of Macrophage Activation. J. Biol. Chem. 2013, 288, 3897–3906.
- Dobrovolskaia, M.A.; Vogel, S.N. Toll Receptors, CD14, and Macrophage Activation and Deactivation by LPS. Microbes Infect. 2002, 4, 903–914.
- Foster, S.L.; Medzhitov, R. Gene-Specific Control of the TLR-Induced Inflammatory Response. Clin. Immunol. 2009, 130, 7–15.
- Hoogendijk, A.J.; Garcia-Laorden, M.I.; Van Vught, L.A.; Wiewel, M.A.; Belkasim-Bohoudi, H.; Duitman, J.; Horn, J.; Schultz, M.J.; Scicluna, B.P.; Van ‘T Veer, C.; et al. Sepsis Patients Display a Reduced Capacity to Activate Nuclear Factor-κB in Multiple Cell Types*. Crit. Care Med. 2017, 45, e524–e531.
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.-S.; et al. PD-1 Expression by Macrophages Plays a Pathologic Role in Altering Microbial Clearance and the Innate Inflammatory Response to Sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308.
- Zhou, M.; Aziz, M.; Yen, H.-T.; Ma, G.; Murao, A.; Wang, P. Extracellular CIRP Dysregulates Macrophage Bacterial Phagocytosis in Sepsis. Cell Mol. Immunol. 2022, 20, 80–93.
- Chen, W.; Qiang, X.; Wang, Y.; Zhu, S.; Li, J.; Babaev, A.; Yang, H.; Gong, J.; Becker, L.; Wang, P.; et al. Identification of Tetranectin-Targeting Monoclonal Antibodies to Treat Potentially Lethal Sepsis. Sci. Transl. Med. 2020, 12, eaaz3833.
- Yang, G.; Zhang, Q.; Tan, J.; Xiong, Y.; Liang, Y.; Yan, J.; Gu, F.; Xu, Y. HMGB1 Induces Macrophage Pyroptosis in Chronic Endometritis. Int. Immunopharmacol. 2023, 123, 110706.
- Zhang, W.; Jiang, H.; Wu, G.; Huang, P.; Wang, H.; An, H.; Liu, S.; Zhang, W. The Pathogenesis and Potential Therapeutic Targets in Sepsis. MedComm 2023, 4, e418.
- Wang, Y.; Zhang, W.; Xu, Y.; Wu, D.; Gao, Z.; Zhou, J.; Qian, H.; He, B.; Wang, G. Extracellular HMGB1 Impairs Macrophage-Mediated Efferocytosis by Suppressing the Rab43-Controlled Cell Surface Transport of CD91. Front. Immunol. 2022, 13, 767630.
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 Enhances Immune Suppression by Facilitating the Differentiation and Suppressive Activity of Myeloid-Derived Suppressor Cells. Cancer Res. 2014, 74, 5723–5733.
- Döcke, W.-D.; Randow, F.; Syrbe, U.; Krausch, D.; Asadullah, K.; Reinke, P.; Volk, H.-D.; Kox, W. Monocyte Deactivation in Septic Patients: Restoration by IFN-γ Treatment. Nat. Med. 1997, 3, 678–681.
- Kim, E.Y.; Ner-Gaon, H.; Varon, J.; Cullen, A.M.; Guo, J.; Choi, J.; Barragan-Bradford, D.; Higuera, A.; Pinilla-Vera, M.; Short, S.A.P.; et al. Post-Sepsis Immunosuppression Depends on NKT Cell Regulation of mTOR/IFN-γ in NK Cells. J. Clin. Investig. 2020, 130, 3238–3252.
- Shen, S.; Shao, Y.; Li, C. Different Types of Cell Death and Their Shift in Shaping Disease. Cell Death Discov. 2023, 9, 284.
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, Ferroptosis, Pyroptosis, and Necroptosis in Tumor Immunotherapy. Sig Transduct. Target. Ther. 2022, 7, 196.
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell Mol. Immunol. 2021, 18, 1106–1121.
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364.
- Rathmell, J.C.; Thompson, C.B. Pathways of Apoptosis in Lymphocyte Development, Homeostasis, and Disease. Cell 2002, 109, S97–S107.
- Alberts, B. (Ed.) Molecular Biology of the Cell, 4th ed.; Garland: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3.
- Tsujimoto, Y. Role of Bcl-2 Family Proteins in Apoptosis: Apoptosomes or Mitochondria?: Role of Bcl-2 Family Proteins in Apoptosis. Genes. Cells 1998, 3, 697–707.
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431.
- Sheikh Motahar Vahedi, H.; Bagheri, A.; Jahanshir, A.; Seyedhosseini, J.; Vahidi, E. Association of Lymphopenia with Short Term Outcomes of Sepsis Patients; a Brief Report. Arch. Acad. Emerg. Med. 2019, 7, e14.
- Carrero, J.; Unanue, E. Lymphocyte Apoptosis as an Immune Subversion Strategy of Microbial Pathogens. Trends Immunol. 2006, 27, 497–503.
- Van Hauwermeiren, F.; Van Opdenbosch, N.; Van Gorp, H.; De Vasconcelos, N.; Van Loo, G.; Vandenabeele, P.; Kanneganti, T.-D.; Lamkanfi, M. Bacillus Anthracis Induces NLRP3 Inflammasome Activation and Caspase-8–Mediated Apoptosis of Macrophages to Promote Lethal Anthrax. Proc. Natl. Acad. Sci. USA 2022, 119, e2116415119.
- Hotchkiss, R.S.; Swanson, P.E.; Knudson, C.M.; Chang, K.C.; Cobb, J.P.; Osborne, D.F.; Zollner, K.M.; Buchman, T.G.; Korsmeyer, S.J.; Karl, I.E. Overexpression of Bcl-2 in Transgenic Mice Decreases Apoptosis and Improves Survival in Sepsis. J. Immunol. 1999, 162, 4148–4156.
- Wang, S.D.; Huang, K.J.; Lin, Y.S.; Lei, H.Y. Sepsis-Induced Apoptosis of the Thymocytes in Mice. J. Immunol. 1994, 152, 5014–5021.
- Luan, Y.; Yao, Y.; Xiao, X.; Sheng, Z. Insights into the Apoptotic Death of Immune Cells in Sepsis. J. Interferon Cytokine Res. 2015, 35, 17–22.
- Luan, Y.-Y.; Dong, N.; Xie, M.; Xiao, X.-Z.; Yao, Y.-M. The Significance and Regulatory Mechanisms of Innate Immune Cells in the Development of Sepsis. J. Interferon Cytokine Res. 2014, 34, 2–15.
- Matsuda, N.; Teramae, H.; Futatsugi, M.; Takano, K.; Yamamoto, S.; Tomita, K.; Suzuki, T.; Yokoo, H.; Koike, K.; Hattori, Y. Up-Regulation of Histamine H 4 Receptors Contributes to Splenic Apoptosis in Septic Mice: Counteraction of the Antiapoptotic Action of Nuclear Factor-κB. J. Pharmacol. Exp. Ther. 2010, 332, 730–737.
- Bo, L.; Wang, F.; Zhu, J.; Li, J.; Deng, X. Granulocyte-Colony Stimulating Factor (G-CSF) and Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) for Sepsis: A Meta-Analysis. Crit. Care 2011, 15, R58.
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmelé, T.; Blood, T.; Morre, M.; et al. Interleukin-7 Restores Lymphocytes in Septic Shock: The IRIS-7 Randomized Clinical Trial. JCI Insight 2018, 3, e98960.
- Zhang, W.; Fang, X.; Gao, C.; Song, C.; He, Y.; Zhou, T.; Yang, X.; Shang, Y.; Xu, J. MDSCs in Sepsis-Induced Immunosuppression and Its Potential Therapeutic Targets. Cytokine Growth Factor. Rev. 2023, 69, 90–103.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Goldmann, O.; Beineke, A.; Medina, E. Identification of a Novel Subset of Myeloid-Derived Suppressor Cells During Chronic Staphylococcal Infection That Resembles Immature Eosinophils. J. Infect. Dis. 2017, 216, 1444–1451.
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150.
- Talmadge, J.E.; Gabrilovich, D.I. History of Myeloid-Derived Suppressor Cells. Nat. Rev. Cancer 2013, 13, 739–752.
- Brudecki, L.; Ferguson, D.A.; McCall, C.E.; El Gazzar, M. Myeloid-Derived Suppressor Cells Evolve during Sepsis and Can Enhance or Attenuate the Systemic Inflammatory Response. Infect. Immun. 2012, 80, 2026–2034.
- Cuenca, A.G.; Delano, M.J.; Kelly-Scumpia, K.M.; Moreno, C.; Scumpia, P.O.; LaFace, D.M.; Heyworth, P.G.; Efron, P.A.; Moldawer, L.L. A Paradoxical Role for Myeloid-Derived Suppressor Cells in Sepsis and Trauma. Mol. Med. 2011, 17, 281–292.
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.L.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-Dependent Expansion of an Immature GR-1+CD11b+ Population Induces T Cell Suppression and Th2 Polarization in Sepsis. J. Exp. Med. 2007, 204, 1463–1474.
- Mathias, B.; Delmas, A.L.; Ozrazgat-Baslanti, T.; Vanzant, E.L.; Szpila, B.E.; Mohr, A.M.; Moore, F.A.; Brakenridge, S.C.; Brumback, B.A.; Moldawer, L.L.; et al. Human Myeloid-Derived Suppressor Cells Are Associated with Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann. Surg. 2017, 265, 827–834.
- Landoni, V.I.; Martire-Greco, D.; Rodriguez-Rodrigues, N.; Chiarella, P.; Schierloh, P.; Isturiz, M.A.; Fernández, G.C. Immature Myeloid Gr-1+ CD11b+ Cells from Lipopolysaccharide-Immunosuppressed Mice Acquire Inhibitory Activity in the Bone Marrow and Migrate to Lymph Nodes to Exert Their Suppressive Function. Clin. Sci. 2016, 130, 259–271.
- Tang, H.; Li, H.; Sun, Z. Targeting Myeloid-Derived Suppressor Cells for Cancer Therapy. Cancer Biol. Med. 2021, 18, 992–1009.
- Uhel, F.; Azzaoui, I.; Grégoire, M.; Pangault, C.; Dulong, J.; Tadié, J.-M.; Gacouin, A.; Camus, C.; Cynober, L.; Fest, T.; et al. Early Expansion of Circulating Granulocytic Myeloid-Derived Suppressor Cells Predicts Development of Nosocomial Infections in Patients with Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 315–327.
- Arocena, A.R.; Onofrio, L.I.; Pellegrini, A.V.; Carrera Silva, A.E.; Paroli, A.; Cano, R.C.; Aoki, M.P.; Gea, S. Myeloid-derived Suppressor Cells Are Key Players in the Resolution of Inflammation during a Model of Acute Infection. Eur. J. Immunol. 2014, 44, 184–194.
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic Acute-Phase Proteins Control Innate Immune Responses during Infection by Promoting Myeloid-Derived Suppressor Cell Function. J. Exp. Med. 2010, 207, 1453–1464.
- Schrijver, I.T.; Karakike, E.; Théroude, C.; Baumgartner, P.; Harari, A.; Giamarellos-Bourboulis, E.J.; Calandra, T.; Roger, T. High Levels of Monocytic Myeloid-Derived Suppressor Cells Are Associated with Favorable Outcome in Patients with Pneumonia and Sepsis with Multi-Organ Failure. Intensive Care Med. Exp. 2022, 10, 5.
- Hollen, M.K.; Stortz, J.A.; Darden, D.; Dirain, M.L.; Nacionales, D.C.; Hawkins, R.B.; Cox, M.C.; Lopez, M.-C.; Rincon, J.C.; Ungaro, R.; et al. Myeloid-Derived Suppressor Cell Function and Epigenetic Expression Evolves over Time after Surgical Sepsis. Crit. Care 2019, 23, 355.