 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dileep Kumar | -- | 5990 | 2024-01-24 08:25:07 | | | |
| 2 | Lindsay Dong | -2 word(s) | 5988 | 2024-01-25 01:09:56 | | |
Video Upload Options
Ongoing therapy for human parasite infections has a few known drugs but with serious side effects and the problem of drug resistance, impelling us to discover novel drug candidates with newer mechanisms of action. Universally, this has boosted the research in the design and development of novel medicinal agents as antiparasitic drugs with a novel mode of action. Histone deacetylase inhibitors (HDACis) are used in a vast variety of diseases due to their anti-inflammatory properties. Drug repurposing strategies have already approved HDACis as cancer therapeutics and are now under investigation for many parasitic infections. Along with the expression of the gene, histone deacetylase (HDAC) enzymes also act as a slice of great multi-subunit complexes, targeting many non-histones, changing systemic and cellular levels signaling, and producing different cell-based specified effects. Zinc (Zn2+)- and nicotinamide adenine dinucleotide (NAD+)-dependent HDACs of parasites play pivotal roles in the alteration of gene expression of parasites. Some of them are already known to be responsible for the survival of several parasites under odd circumstances; thus, targeting them for therapeutic interventions will be novel for potential antiparasitic targets.
1. Introduction
2. Opportunities of HDAC Inhibitors as Antiparasitic Agent
2.1. HDAC Inhibitors as Antimalarial Agent
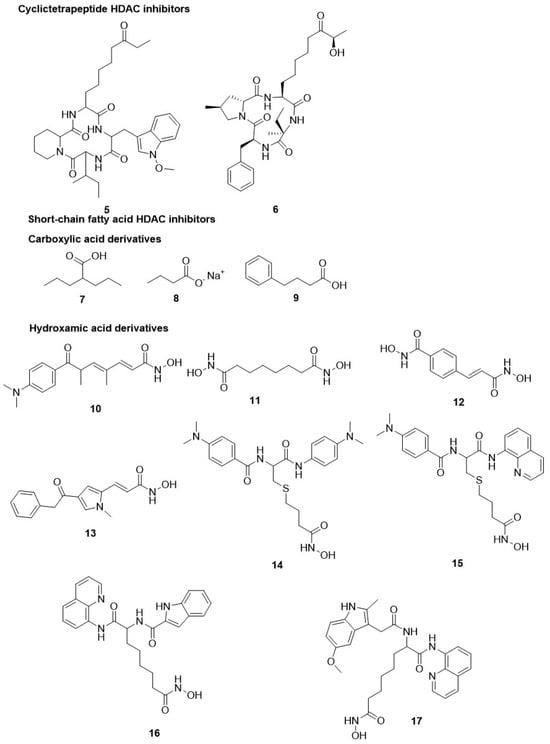
2.1.1. Cyclictetrapeptide HDAC Inhibitors
2.1.2. Short-Chain Fatty Acid HDAC Inhibitors
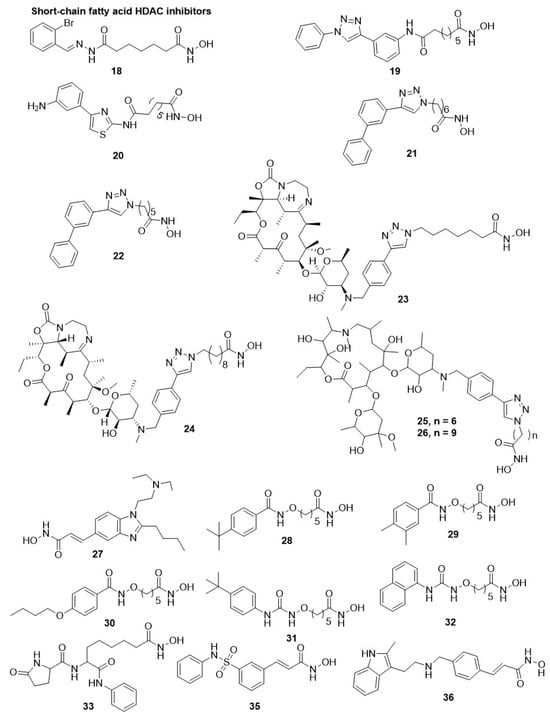
2.1.3. Hydroxamate-Based HDAC Inhibitors
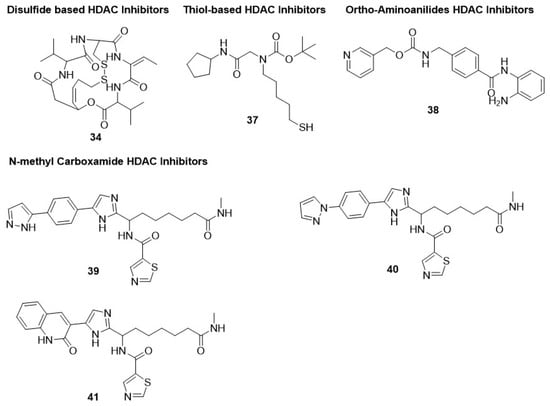
2.1.4. Thiol-Based HDAC Inhibitors
2.1.5. Ortho-Aminoanilides HDAC Inhibitors
2.2. Antimalarial Class-III HDAC (Sirtuin) Inhibitors
2.3. Antitrypanosomal HDAC Inhibitors
2.4. Antitrypanosomal Class-III HDAC (Sirtuin) Inhibitors
2.5. Antileishmanial HDAC Inhibitors
2.6. Antileishmanial Class-III HDAC (Sirtuin) Inhibitors
2.7. Antitoxoplasma HDAC Inhibitors
References
- World Health Organization. World Malaria Report 2022; World Health Organization: Geneva, Switzerland, 2022.
- World Health Organization. World Malaria Report 2014: Summary; World Health Organization: Geneva, Switzerland, 2015.
- Pigott, D.M.; Bhatt, S.; Golding, N.; Duda, K.A.; Battle, K.E.; Brady, O.J.; Messina, J.P.; Balard, Y.; Bastien, P.; Pratlong, F. Global distribution maps of the leishmaniases. eLife 2014, 3, e02851.
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264.
- Nemati Zargaran, F.; Rostamian, M.; Kooti, S.; Madanchi, H.; Ghadiri, K. Co-infection of COVID-19 and parasitic diseases: A systematic review. Parasite Epidemiol. Control 2023, 21, e00299.
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252.
- Labella, D. Design, Synthesis and Biological Evaluation of Novel Epigenetic Modulators; Sapienza University of Rome: Rome, Italy, 2014.
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467.
- Berg, M.; García-Hernández, R.; Cuypers, B.; Vanaerschot, M.; Manzano, J.I.; Poveda, J.A.; Ferragut, J.A.; Castanys, S.; Dujardin, J.-C.; Gamarro, F. Experimental resistance to drug combinations in Leishmania donovani: Metabolic and phenotypic adaptations. Antimicrob. Agents Chemother. 2015, 59, 2242–2255.
- Takala-Harrison, S.; Jacob, C.G.; Arze, C.; Cummings, M.P.; Silva, J.C.; Dondorp, A.M.; Fukuda, M.M.; Hien, T.T.; Mayxay, M.; Noedl, H. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 2015, 211, 670–679.
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77.
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Vert, J.P.; Noble, W.S.; Le Roch, K.G. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. falciparum. Bioessays 2015, 37, 182–194.
- Cheeseman, K.; Weitzman, J.B. Host–parasite interactions: An intimate epigenetic relationship. Cell. Microbiol. 2015, 17, 1121–1132.
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111.
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400.
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112.
- Montenegro, M.; Sanchez-del-Campo, L.; Fernandez-Perez, M.; Saez-Ayala, M.; Cabezas-Herrera, J.; Rodriguez-Lopez, J. Targeting the epigenetic machinery of cancer cells. Oncogene 2015, 34, 135–143.
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336.
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901.
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691.
- Brien, G.L.; Valerio, D.G.; Armstrong, S.A. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell 2016, 29, 464–476.
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer therapy with HDAC inhibitors: Mechanism-based combination strategies and future perspectives. Cancers 2021, 13, 634.
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291.
- Coleman, B.I.; Skillman, K.M.; Jiang, R.H.; Childs, L.M.; Altenhofen, L.M.; Ganter, M.; Leung, Y.; Goldowitz, I.; Kafsack, B.F.; Marti, M. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe 2014, 16, 177–186.
- T Andrews, K.; N Tran, T.; P Fairlie, D. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr. Pharm. Des. 2012, 18, 3467–3479.
- Marek, M.; Oliveira, G.; Pierce, R.J.; Jung, M.; Sippl, W.; Romier, C. Drugging the schistosome zinc-dependent HDACs: Current progress and future perspectives. Future Med. Chem. 2015, 7, 783–800.
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361.
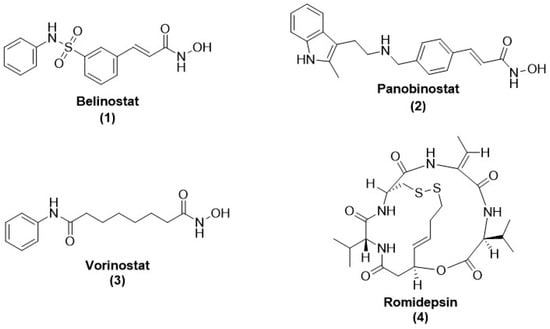
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22.
- Prince, H.M.; Dickinson, M. Romidepsin for Cutaneous T-cell Lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515.
- Thompson, C.A. Belinostat Approved for Use in Treating Rare Lymphoma; Oxford University Press: Oxford, UK, 2014.
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704.
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147.
- Patel, V.; Mazitschek, R.; Coleman, B.; Nguyen, C.; Urgaonkar, S.; Cortese, J.; Barker, R.H., Jr.; Greenberg, E.; Tang, W.; Bradner, J.E. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187.
- Hu, G.; Cabrera, A.; Kono, M.; Mok, S.; Chaal, B.K.; Haase, S.; Engelberg, K.; Cheemadan, S.; Spielmann, T.; Preiser, P.R. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010, 28, 91–98.
- Chaal, B.K.; Gupta, A.P.; Wastuwidyaningtyas, B.D.; Luah, Y.-H.; Bozdech, Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010, 6, e1000737.
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163.
- Peart, M.J.; Smyth, G.K.; Van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702.
- Meinke, P.T.; Colletti, S.L.; Doss, G.; Myers, R.W.; Gurnett, A.M.; Dulski, P.M.; Darkin-Rattray, S.J.; Allocco, J.J.; Galuska, S.; Schmatz, D.M. Synthesis of apicidin-derived quinolone derivatives: Parasite-selective histone deacetylase inhibitors and antiproliferative agents. J. Med. Chem. 2000, 43, 4919–4922.
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 2. Bioorganic Med. Chem. Lett. 2001, 11, 113–117.
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 1. Bioorganic Med. Chem. Lett. 2001, 11, 107–111.
- Murray, P.J.; Kranz, M.; Ladlow, M.; Taylor, S.; Berst, F.; Holmes, A.B.; Keavey, K.N.; Jaxa-Chamiec, A.; Seale, P.W.; Stead, P. The synthesis of cyclic tetrapeptoid analogues of the antiprotozoal natural product apicidin. Bioorganic Med. Chem. Lett. 2001, 11, 773–776.
- Bougdour, A.; Maubon, D.; Baldacci, P.; Ortet, P.; Bastien, O.; Bouillon, A.; Barale, J.-C.; Pelloux, H.; Ménard, R.; Hakimi, M.-A. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med. 2009, 206, 953–966.
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13.
- Strobl, J.S.; Cassell, M.; Mitchell, S.M.; Reilly, C.M.; Lindsay, D.S. Scriptaid and suberoylanilide hydroxamic acid are histone deacetylase inhibitors with potent anti–Toxoplasma gondii activity in vitro. J. Parasitol. 2007, 93, 694–700.
- Jones-Brando, L.; Torrey, E.F.; Yolken, R. Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr. Res. 2003, 62, 237–244.
- Ingram, A.K.; Horn, D. Histone deacetylases in Trypanosoma brucei: Two are essential and another is required for normal cell cycle progression. Mol. Microbiol. 2002, 45, 89–97.
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126.
- Rotili, D.; Simonetti, G.; Savarino, A.; Palamara, A.T.; Migliaccio, A.R.; Mai, A. Non-Cancer Uses of Histone Deacetylase Inhibitors: Effects on Infectious Diseases and β-Hemoglobinopathies. Curr. Top. Med. Chem. 2009, 9, 272–291.
- Dow, G.S.; Chen, Y.; Andrews, K.T.; Caridha, D.; Gerena, L.; Gettayacamin, M.; Johnson, J.; Li, Q.; Melendez, V.; Obaldia III, N. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 3467–3477.
- Andrews, K.T.; Walduck, A.; Kelso, M.J.; Fairlie, D.P.; Saul, A.; Parsons, P.G. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int. J. Parasitol. 2000, 30, 761–768.
- Mai, A.; Cerbara, I.; Valente, S.; Massa, S.; Walker, L.A.; Tekwani, B.L. Antimalarial and antileishmanial activities of aroyl-pyrrolyl-hydroxyamides, a new class of histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2004, 48, 1435.
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309.
- Andrews, K.; Tran, T.; Lucke, A.; Kahnberg, P.; Le, G.; Boyle, G.; Gardiner, D.; Skinner-Adams, T.; Fairlie, D. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461.
- Andrews, K.T.; Gupta, A.P.; Tran, T.N.; Fairlie, D.P.; Gobert, G.N.; Bozdech, Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PLoS ONE 2012, 7, e31847.
- Marfurt, J.; Chalfein, F.; Prayoga, P.; Wabiser, F.; Kenangalem, E.; Piera, K.A.; Fairlie, D.P.; Tjitra, E.; Anstey, N.M.; Andrews, K.T. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 2011, 55, 961–966.
- Wheatley, N.C.; Andrews, K.T.; Tran, T.L.; Lucke, A.J.; Reid, R.C.; Fairlie, D.P. Antimalarial histone deacetylase inhibitors containing cinnamate or NSAID components. Bioorganic Med. Chem. Lett. 2010, 20, 7080–7084.
- Alcaín, F.J.; Villalba, J.M. Sirtuin inhibitors. Expert Opin. Ther. Pat. 2009, 19, 283–294.
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448.
- Agbor-Enoh, S.; Seudieu, C.; Davidson, E.; Dritschilo, A.; Jung, M. Novel inhibitor of Plasmodium histone deacetylase that cures P. berghei-infected mice. Antimicrob. Agents Chemother. 2009, 53, 1727–1734.
- Patil, V.; Guerrant, W.; Chen, P.C.; Gryder, B.; Benicewicz, D.B.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorganic Med. Chem. 2010, 18, 415–425.
- Mwakwari, S.C.; Guerrant, W.; Patil, V.; Khan, S.I.; Tekwani, B.L.; Gurard-Levin, Z.A.; Mrksich, M.; Oyelere, A.K. Non-peptide macrocyclic histone deacetylase inhibitors derived from tricyclic ketolide skeleton. J. Med. Chem. 2010, 53, 6100–6111.
- Guerrant, W.; Mwakwari, S.C.; Chen, P.C.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. A structure–activity relationship study of the antimalarial and antileishmanial activities of nonpeptide macrocyclic histone deacetylase inhibitors. ChemMedChem 2010, 5, 1232–1235.
- Sumanadasa, S.D.; Goodman, C.D.; Lucke, A.J.; Skinner-Adams, T.; Sahama, I.; Haque, A.; Do, T.A.; McFadden, G.I.; Fairlie, D.P.; Andrews, K.T. Antimalarial activity of the anticancer histone deacetylase inhibitor SB939. Antimicrob. Agents Chemother. 2012, 56, 3849–3856.
- Hansen, F.K.; Sumanadasa, S.D.; Stenzel, K.; Duffy, S.; Meister, S.; Marek, L.; Schmetter, R.; Kuna, K.; Hamacher, A.; Mordmüller, B. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014, 82, 204–213.
- Hansen, F.K.; Skinner-Adams, T.S.; Duffy, S.; Marek, L.; Sumanadasa, S.D.; Kuna, K.; Held, J.; Avery, V.M.; Andrews, K.T.; Kurz, T. Synthesis, Antimalarial Properties, and SAR Studies of Alkoxyurea-Based HDAC Inhibitors. ChemMedChem 2014, 9, 665–670.
- Trenholme, K.; Marek, L.; Duffy, S.; Pradel, G.; Fisher, G.; Hansen, F.K.; Skinner-Adams, T.S.; Butterworth, A.; Ngwa, C.J.; Moecking, J. Lysine acetylation in sexual stage malaria parasites is a target for antimalarial small molecules. Antimicrob. Agents Chemother. 2014, 58, 3666–3678.
- Giannini, G.; Battistuzzi, G.; Vignola, D. Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorganic Med. Chem. Lett. 2015, 25, 459–461.
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure−selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438.
- Prusty, D.; Mehra, P.; Srivastava, S.; Shivange, A.V.; Gupta, A.; Roy, N.; Dhar, S.K. Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth. FEMS Microbiol. Lett. 2008, 282, 266–272.
- Chakrabarty, S.P.; Saikumari, Y.K.; Bopanna, M.P.; Balaram, H. Biochemical characterization of Plasmodium falciparum Sir2, a NAD+-dependent deacetylase. Mol. Biochem. Parasitol. 2008, 158, 139–151.
- Tonkin, C.J.; Carret, C.K.; Duraisingh, M.T.; Voss, T.S.; Ralph, S.A.; Hommel, M.; Duffy, M.F.; Silva, L.M.d.; Scherf, A.; Ivens, A. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009, 7, e1000084.
- Duraisingh, M.T.; Voss, T.S.; Marty, A.J.; Duffy, M.F.; Good, R.T.; Thompson, J.K.; Freitas-Junior, L.H.; Scherf, A.; Crabb, B.S.; Cowman, A.F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24.
- Verotta, L.; Appendino, G.; Bombardelli, E.; Brun, R. In vitro antimalarial activity of hyperforin, a prenylated acylphloroglucinol. A structure–activity study. Bioorganic Med. Chem. Lett. 2007, 17, 1544–1548.
- Merrick, C.J.; Duraisingh, M.T. Plasmodium falciparum Sir2: An unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot. Cell 2007, 6, 2081–2091.
- Gey, C.; Kyrylenko, S.; Hennig, L.; Nguyen, L.H.D.; Büttner, A.; Pham, H.D.; Giannis, A. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: Inhibitors of human sirtuins SIRT1 and SIRT2. Angew. Chem. Int. Ed. 2007, 46, 5219–5222.
- Chakrabarty, S.P.; Ramapanicker, R.; Mishra, R.; Chandrasekaran, S.; Balaram, H. Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity. Bioorganic Med. Chem. 2009, 17, 8060–8072.
- Sheader, K.; te Vruchte, D.; Rudenko, G. Bloodstream form-specific up-regulation of silent vsg expression sites and procyclin in Trypanosoma brucei after inhibition of DNA synthesis or DNA damage. J. Biol. Chem. 2004, 279, 13363–13374.
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61.
- Respuela, P.; Ferella, M.; Rada-Iglesias, A.; Åslund, L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J. Biol. Chem. 2008, 283, 15884–15892.
- Kelly, J.M.; Taylor, M.C.; Horn, D.; Loza, E.; Kalvinsh, I.; Björkling, F. Inhibitors of human histone deacetylase with potent activity against the African trypanosome Trypanosoma brucei. Bioorganic Med. Chem. Lett. 2012, 22, 1886–1890.
- Carrillo, A.K.; Guiguemde, W.A.; Guy, R.K. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorganic Med. Chem. 2015, 23, 5151–5155.
- Soares, M.B.; Silva, C.V.; Bastos, T.M.; Guimarães, E.T.; Figueira, C.P.; Smirlis, D.; Azevedo, W.F., Jr. Anti-Trypanosoma cruzi activity of nicotinamide. Acta Trop. 2012, 122, 224–229.
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791.
- Moretti, N.S.; da Silva Augusto, L.; Clemente, T.M.; Antunes, R.P.P.; Yoshida, N.; Torrecilhas, A.C.; Cano, M.I.N.; Schenkman, S. Characterization of Trypanosoma cruzi sirtuins as possible drug targets for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 4669–4679.
- Rotili, D.; Tarantino, D.; Nebbioso, A.; Paolini, C.; Huidobro, C.; Lara, E.; Mellini, P.; Lenoci, A.; Pezzi, R.; Botta, G. Discovery of salermide-related sirtuin inhibitors: Binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J. Med. Chem. 2012, 55, 10937–10947.
- Sacconnay, L.; Smirlis, D.; Queiroz, E.F.; Wolfender, J.L.; Soares, M.B.P.; Carrupt, P.-A.; Nurisso, A. Structural insights of SIR2rp3 proteins as promising biotargets to fight against Chagas disease and leishmaniasis. Mol. BioSyst. 2013, 9, 2223–2230.
- Vergnes, B.; Vanhille, L.; Ouaissi, A.; Sereno, D. Stage-specific antileishmanial activity of an inhibitor of SIR2 histone deacetylase. Acta Trop. 2005, 94, 107–115.
- Sodji, Q.; Patil, V.; Jain, S.; Kornacki, J.R.; Mrksich, M.; Tekwani, B.L.; Oyelere, A.K. The antileishmanial activity of isoforms 6-and 8-selective histone deacetylase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 4826–4830.
- Sereno, D.; Alegre, A.M.; Silvestre, R.; Vergnes, B.; Ouaissi, A. In vitro antileishmanial activity of nicotinamide. Antimicrob. Agents Chemother. 2005, 49, 808–812.
- Tavares, J.; Ouaissi, A.; Silva, A.M.; Lin, P.K.T.; Roy, N.; Cordeiro-da-Silva, A. Anti-leishmanial activity of the bisnaphthalimidopropyl derivatives. Parasitol. Int. 2012, 61, 360–363.
- Theurkauf, W.E.; Baum, H.; Bo, J.; Wensink, P.C. Tissue-specific and constitutive alpha-tubulin genes of Drosophila melanogaster code for structurally distinct proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 8477–8481.
- Kadam, R.U.; Tavares, J.; Cordeiro, A.; Ouaissi, A.; Roy, N. Structure function analysis of Leishmania sirtuin: An ensemble of in silico and biochemical studies. Chem. Biol. Drug Des. 2008, 71, 501–506.
- Tavares, J.; Ouaissi, A.; Kong Thoo Lin, P.; Loureiro, I.; Kaur, S.; Roy, N.; Cordeiro-da-Silva, A. Bisnaphthalimidopropyl derivatives as inhibitors of Leishmania SIR2 related protein 1. ChemMedChem Chem. Enabling Drug Discov. 2010, 5, 140–147.
- Maubon, D.; Bougdour, A.; Wong, Y.-S.; Brenier-Pinchart, M.-P.; Curt, A.; Hakimi, M.-A.; Pelloux, H. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: Inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob. Agents Chemother. 2010, 54, 4843–4850.
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.-T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from Schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435.
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Langley, B.C. Functional differences in epigenetic modulators superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061.

