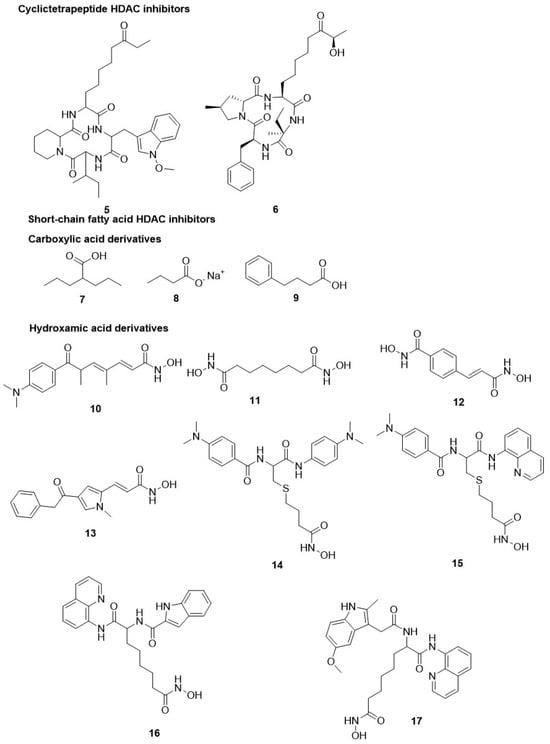
Patel et al. performed a high-throughput assay to study the antimalarial efficacy of approximately 2000 HDACis obtained from the compound library. The library was characterized by a moiety of acyl hydrazone as CU and with diverse moieties for CAP protein and zinc-binding group along with the hydrophobic linker length such as 4–6 methylene units [
81]. The study revealed that numerous compounds strongly inhibited the growth of
P. falciparum and also hampered recombinant PfHDAC1 enzymatic activity. Among them, seventeen derivatives displayed a low range of nanomolar antiparasitic activity for acetylation of histone along with minimal perturbation of human myeloma MM1S cells as an indicator of selectivity [
95]. Within this series, the selective inhibition of
P. falciparum growth was highly enhanced by the existence of ortho-substituents, such as bromine and hydroxyl, in the aromatic group of CAP protein along with the presence of metal chelator or hydroxamic acid and five methylene units as a linker (compound
18,
Table 1) [
95]. Another study by Kozikowski et al. on two series of suberoyl amide hydroxamates substituted moieties of triazolyl phenyl and phenyl thiazolyl as CAP protein groups [
137,
143]. The triazolyl phenyl-based compound
19 showed high efficacy against the multidrug-resistant strains of
P. falciparum (C2A and C235), with IC
50 values in a range of 0.017–0.035 µM. It was 10-fold more potent compared to its counter congeners such as chloroquine and mefloquine. Compound
19 was 23-fold highly selective for C235 over mammalian cells [
143]. In a group of 50 phenyl, thiazolyl hydroxamate-based HDACi, three compounds were highly potent with IC
50 values < 3 nM and showed 600-fold selectivity against
P. falciparum compared to mammalian cells. Compound
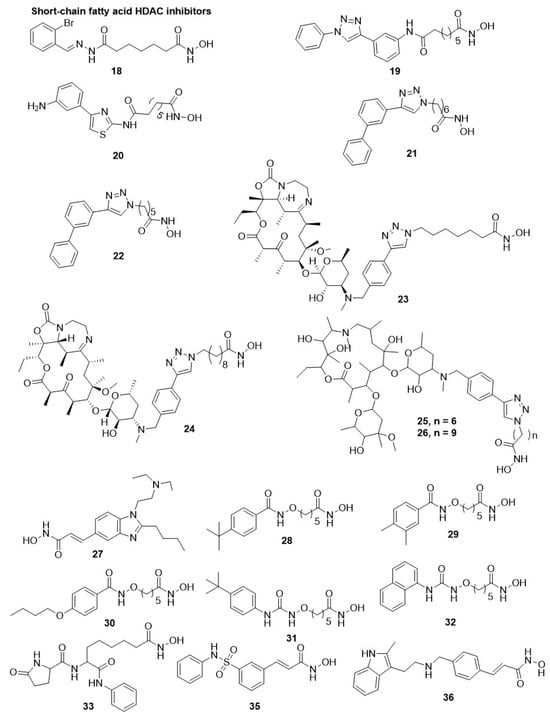
20 (WR301801) was the most favorable HDACi derivative, with IC
50 values 0.6–16 nM for several drug-resistant strains, D6, W2, C235, and C2A, of
P. falciparum. It displayed notable inhibition of HDAC activity in nuclear extracts of
P. falciparum with IC
50 ∼10 nM along with a strong in situ parasitic histones hyperacetylation [
137]. In another report, compound
20, upon oral administration as monotherapy, at doses of up to 640 mg/kg demonstrated a notable suppression of parasitemia but did not rehabilitate
P. berghei-infected mice. However, some mice, but not all mice, were rehabilitated upon treatment with compound
20 (52 mg/kg) when co-administered with a subcurative dose of chloroquine (64 mg/kg) [
137]. Similarly, compound
20 on oral administration at a dose of 32 mg/kg/day for 3 days to Aotus monkeys infected with
P. falciparum resulted in suppression of the parasite but not complete elimination [
137]. Another reported study showed that compound
20 (50 mg/kg/day for 4 days) on i.p. injection with an experimental follow-up period of 6 weeks enhanced survival of infected mice infected with
P. berghei and irreversibly downregulated the parasitemia [
144]. However, the optimization of the pharmacokinetic properties of compound
20 would show advantageous results as it is quickly hydrolyzed to the corresponding inactive carboxylic acid [
137]. These findings suggest the potential of HDACi in mono/combination therapy for malaria treatment [
12,
26,
137,
144]. Oyelere et al., reported a series of aryl triazolyl hydroxamate-based HDACis that were screened for their inhibitory activity against
L. donovani promastigote stages and asexual blood stage of
P. falciparum [
145]. Several compounds displayed improved inhibitory activity and selectivity than compound
3 against D6 and W2 strains of
P. falciparum. Although several compounds are less active against
P. falciparum, they showed a modest potency of inhibition against
L. donovani, with 2- to 4-fold better IC
50 values than compound
3 and equivalent to the standard drug miltefosine, used to treat visceral Leishmaniasis. Antiparasitic activity depends on polymethylene linker length and the nature of the CAP group. The CAP moiety showed maximum activity against both parasites when 5 and 6 methylene units were present in the spacer region between the CAP and the ZBG. Compounds
21 and
22, with a CAP protein (3′-biphenyltriazolyl moiety) and linker (6 and 5 methylene units), showed the highest selectivity and activity against
P. falciparum. Compound
22 also showed its activity against
L. donovani with an IC
50 of 32 µM [
145]. Based upon this concept, Oyelere et al., tested five tricyclic ketolide-based phenyltriazolyl HDACi for antimalarial and antileishmanial activities [
146]. During the study, it was noticed that compound
23, with six methylene units as the optimal linker present between the CAP and hydroxamic acid, displayed optimal antimalarial activity. Similarly, compound
24, hydroxamic acid with nine methylene units, displayed the best antileishmanial activity, but the study does not correlate with the PfHDAC1 inhibition [
146]. Particularly, compound
23 displayed IC
50 in the range of 0.144–0.148 µM against both chloroquine-sensitive (D6) and chloroquine-resistant (W2)
P. falciparum strains. However, the compound displayed 7 to 10-fold lesser activity than compound
3 and 10-fold high selectivity against mammalian Vero cells without antileishmanial activity. Further, compound
24 showed anti-
L. donovani activity with an IC
50 of 5 µM, which was 16-fold stronger than compound
3 with an IC
50 of 81 µM [
146] (
Figure 4).
Figure 4. Chemical structure of hydroxamic acid HDAC inhibitors. The short chain fatty acid displays diverse HDAC inhibition.
Oyelere et al., further investigated and described the antimalarial and antileishmanial potential of 14 and 15-membered nonpeptide macrocyclics connected to a moiety of phenyltriazolyl, a CAP protein group, as HDACis. All compounds inhibited the proliferation of chloroquine-sensitive (D6) and chloroquine-resistant (W2) strains of
P. falciparum [
155]. However, the best activity and selectivity against
P. falciparum were attained for the six methylene units linker between CAP (triazole ring) and ZBG group (hydroxamate). Among them, compound
25, a skeleton of 15-membered macrolide with an IC
50 value of 29 nM against PfHDAC1 enzyme, manifested 11-fold higher antiplasmodial activity with 14-fold enhanced selectivity over mammalian Vero cells compared to compound
3. Surprisingly, the linker group having five to seven methylene units was lacking anti-
L. donovani activity on the promastigote stage for both skeletons of a macrolide, whereas the compound having eight or nine methylene units as linker displayed the highest activity similar to ketolide-based HDACis, but distinct from the structure–activity relationship was noticed in aryl triazolyl hydroxamates [
145].
Specifically, compound
26 with a skeleton of 14-membered macrolide and methylene units linker was 25-fold more active than compound
3 with an IC
50 value of 81 µM, showing the best antileishmanial activity, with IC
50 values of 3.2 and 4.7 µM against promastigote and amastigote stages of the parasite, respectively [
155]. Andrews et al., identified compound
27 (pracinostat or SB939), an orally active anticancer HDACi, having antiplasmodial activities [
156]. Furthermore, compound
27 strongly hampered the proliferation of the asexual-stage of
P. falciparum in mammalian erythrocytes, with IC
50 values in the range of 0.08–0.15 µM. Compound
27 also caused hyperacetylation on histone and non-histone proteins in the parasite and displayed selectivity over mammalian cells ranging 4 to >1250-fold over tested cell lines [
156]. Additionally, an additive effect was first observed for its combination with lopinavir, a protease inhibitor, as antimalarial targeting a liver stage. Compounds
27 strongly inhibit the exo-erythrocytic stage of
P. berghei in human HepG2hepatocytes, with an IC
50 value of 150 nM [
156]. The oral administration of compound
27 up to a dose of 100 mg/kg/day decreased peripheral parasitemia and total parasite load in the ANKA mouse model infected with
P. berghei. However, the administration of compound
27 to mice fended off the occurrence of cerebral malaria-like symptoms up to 2–3 weeks after the intervention but did not affect hyperparasitemia in the treated animals [
156]. Oyelere et al. also explored a group of 21 HDACi with Pentyloxyamide as a linker and substituted benzene ring as CAP for their effect against different stages of
Plasmodium, such as
P. falciparum (a sexual blood stage (3D7) cell line),
P. berghi (tissue schizontocidal stage), and late stage of
P. falciparum gametocyte (IV and V). All compounds exhibited antimalarial activity against
P. falciparum asexual form with potency and selectivity enhanced as the bulkiness of alkyl/alkoxy substituents in the phenyl ring at the
para position were increased regarding compound
28. Three derivatives, compounds
28,
29, and
30, showed their activity against all three stages of
P. falciparum with IC
50 values of 0.09, 0.12, and 0.17 µM, respectively. Several compounds of this series displayed better selectivity against the parasite compared to compound
3 for the asexual and exo-erythrocytic life cycle stages of the parasite [
157]. Further, Oyelere et al., described the structure–activity relationship and antimalarial activity of a group of HDACi based on alkoxyurea. Some compounds actively inhibited
P. falciparum (3D7cell line) and showed gametocytocidal activities at early and late-developmental stages with IC
50 values ranging 1.68–6.65 µM [
147,
148]. Structure–activity relationship study revealed that the hydroxamic acid as a zinc-binding group and 5 methylene units as a linker were important for antiplasmodial activity N-methyl hydroxamic acid, o-hydroxyanilide and o-amino anilide were inactive as a zinc-binding group, while the short-chain analogues with linker less than 5 methylene units displayed lower potency [
147]. Furthermore, the 4th position is substituted with bulky alkyl/alkoxy substituent of the phenyl ring of the CAP group. When this group is replaced with bulky aromatic rings leads to the development of more potent and effective compounds against asexual and gametocyte forms of
P. falciparum, as denoted by the 4-tert-butyl derivative (
31) and 1-naphthyl derivative (
32). However, compounds
31 and
32 did not display better potency than compound
3, but, under the test condition,
32 displayed higher selectivity than compound
3 along with gametocidal activity in the asexual and later stages [
148].
In 2015, Giannini et al., evaluated twelve analogues for their antiparasitic activity against
P. falciparum,
T. cruzi,
L. donovani,
G. lamblia, and
T. brucei. These twelve analogues were differentiated at the α position of the anilide (CU) and meta position of the phenyl ring (CAP) and substituted with β-lactam-carboxamides and trifluoro methyl groups, respectively. Additionally, a thiol or hydroxamic acid is present as a zinc-binding group. Compound
33 of this series displayed a significant IC
50 of 0.019 µM against
P. falciparum [
149]. In 2015, Andrews et al., described the potential of four clinically approved HDACi, compounds
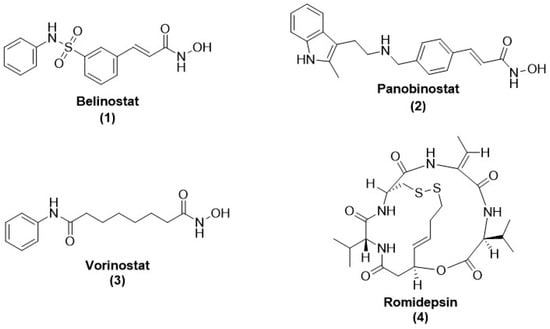
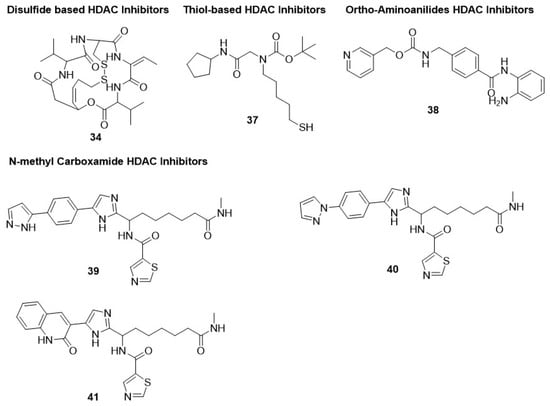
34 (romidepsin or FK228),
35 (belinostat),
36 (panobinostat), and
3, for cancer treatment against
P. falciparum and
T. brucei parasites. All compounds inhibited the growth of
P. falciparum parasite, with IC
50 values ranging from 0.09–0.13, 0.025–2.2, 0.06–0.13, and 0.01–0.03 µM, respectively. Interestingly only compound
34 was active against the bloodstream form of
T. brucei, with an IC
50 value of 35 nM, despite lacking mammalian cell selectivity. These four HDACi inhibited the
P. falciparum nuclear extract deacetylase activity and recombinant PfHDAC1 due to hyperacetylation of non-histone protein and contrastingly affecting histones (H3 and H4) acetylation. Compounds
3 and
35 did not show selectivity towards malaria parasites over mammalian HEK29 and NFF 3 cells [
148] (
Figure 5).
Figure 5. Chemical structure of disulfide-based, thiol-based, orthoamino-anilide-based, and N-methylcarboxamide-based HDAC inhibitors. These inhibitors displayed anti-malarial activity.
2.1.4. Thiol-Based HDAC Inhibitors
Thiol-based HDAC6-selective inhibitor
37 displayed poor inhibitory potential against chloroquine-sensitive (3D7) and chloroquine-resistant (Dd2) strains of
P. falciparum, with IC
50 values in the range of 15.2–19.9 µM [
150]. Thus, further verification is needed to validate the antimalarial potential of hydroxamate-based pan-HDACi [
93] (
Figure 5).
2.1.5. Ortho-Aminoanilides HDAC Inhibitors
A similar inference can be made by choosing an
ortho-amino anilide group as a zinc-binding group. The precursor of these HDACi in the class-I-HDAC-selective inhibitor is denoted as compound
38 (MS275 or entinostat) [
154,
155,
156]. Multiple trials have been completed on compound
38, revealing its potential as a PfHDAC1 inhibitor and parasitic proliferation inhibitor, with IC
50 values of 0.94 and 8 µM, respectively [
12,
26,
88,
93,
95]. Compounds
39,
40, and
41 are N-methyl carboxamide derivatives that displayed antimalarial potential against
P. falciparum [
158] (
Figure 5).
2.2. Antimalarial Class-III HDAC (Sirtuin) Inhibitors
Fewer Sir2 inhibitors have been tested against
P. falciparum-infected erythrocytes for their antiproliferative activity and inhibition of the recombinant PfSir2A protein [
12,
26]. Generally, the Sir2 inhibitors showed moderate activity against the growth of
P. falciparum. A poor homology has been observed between the parasite and other eukaryotic Sir2 proteins [
97,
98]. Several known natural and synthetic sirtuin inhibitors that have been investigated over the years for growth inhibition of
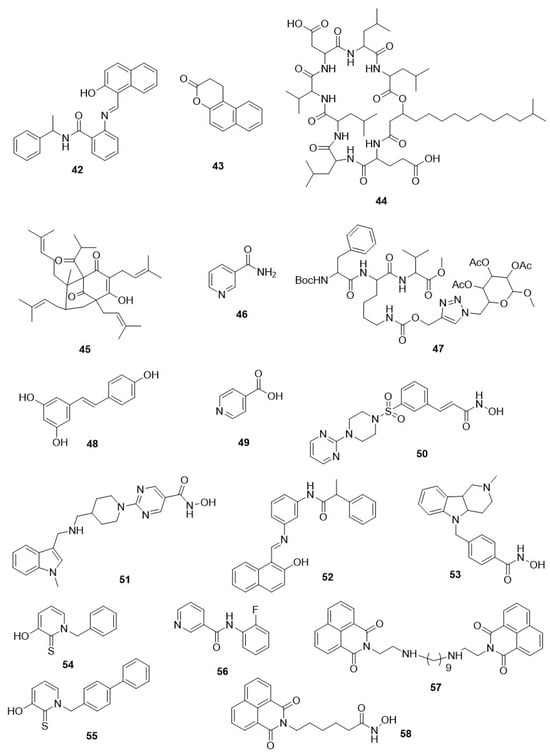
P. falciparum. Compound
42 (sirtinol),
43 (splitomycin),
44 (surfactin), and
45 (hyperforin) displayed IC
50 values in the range of 9–13, >10, 9, and 1.5–2 µM, respectively, against
P. falciparum [
153,
154,
159,
160]. Compound
46 (nicotinamide) is a Sir2-catalysed reaction product and causes non-competitive inhibition of both acetylated peptide and NAD
+ but less active at the whole-cell level, with an IC
50 value of 9.9 mM for retarding parasite growth [
161]. Nicotinic acid, also a product of Sir2-dependent enzymatic pathways, did not show this effect [
161].
Compounds
44 and
46 displayed different activity against PfSir2A, with IC
50 values of 35 and 51 µM, respectively. Meanwhile, compounds
43 and
44 showed less active activity, with IC
50 values of >400 and >50 µM, respectively. Chakrabarty et al., synthesized analogues of lysine-based tripeptide based on a mechanism of competition with the binding pocket of PfSir2 [
162]. From the four analogues, three analogues showed equivalent or improved activity, with IC
50 values in the range of 23–34 µM against PfSir2A in contrast to compounds
44 and
46 [
162]. Compound
47 was highly potent against PfSir2A and also tested against
P. falciparum-infected erythrocytes where it displayed parasitic inhibitory potential similar to compounds
34 and
36 with an IC
50 value of 9.8 µM [
162]. Resveratrol (
48) and isonicotinic acid (
49) are activators of hSIRT1 that moderately inhibit
P. falciparum growth, but during in vitro testing against recombinant PfSir2A, no enzymatic activation or inhibition was reported [
79,
160]. However, this is fully anticipated as PfSir2A and PfSir2B are not required for the growth and development of the parasite and share a few homological sequences with Sir2 proteins of other eukaryotes [
97,
98]. In addition, several compounds,
42,
43,
46, and, to a lesser extent, compound
44, displayed lower inhibitory potential against PfSir2A and the in vitro antiproliferative effects due to their multiple unrelated characters at a single gene locus. Thus, the identification and development of inhibitors with a notable enhancement of potency against PfSir2 and the selectivity towards sirtuins of humans could be a functional gadget considering
P. falciparum sirtuins’ biology and their pharmacological validation as a target of drugs that either directly or indirectly produce antiparasitic activity through obstructing the parasite avoidance from the host innate immune system [
97,
98] (
Figure 5).
2.3. Antitrypanosomal HDAC Inhibitors
A few compounds were tested against the
Trypanosoma species. Compound
10 appeared to prevent the spread of the bloodstream form of
T. brucei at a concentration of 7 µM but without affecting a silent variant surface glycoprotein expression site [
163]. Another study was conducted where the similar compound
10 hampered recombinant TbDAC3 and TbDAC1 activity by >50% at 5 µM concentration. However, this compound cannot alter the acetylation of the histone protein of the parasite at a concentration of 0.3 µM [
146]. Further, four HDACis, compounds
34,
3,
35, and
36, were also tested by Engel et al., for the inhibition of trypanosomal and malaria parasites [
164]. The result of the study revealed that all inhibitors effectively inhibit
T. brucei but all of them displayed cytotoxicity towards the human NFF and HEK 293 cells at lower concentrations, with a selectivity index less than 1 [
136]. However, compound
34 is highly potent against
T. brucei with an IC
50 value of 35 µM.
In 2012, several clinically approved hydroxymates-based HDACis like long-chain amides, sulphonamides, heterocycle-containing acrylamides, and sulphonylpiperazines were tested against a cultured bloodstream form of
T. brucei for their inhibitory effect [
165]. The result of the study indicated that long-chain amides with IC
50 values > 10 µM displayed a notable inhibitory property on the growth in the range of IC
50 values 0.034–1.54 µM. Among them, sulphonylpiperazines with heteroaryl ring attached to the piperazine moiety displayed potent inhibition. Compound
50 displayed an IC
50 value of 34 µM and was able to induce parasite death at 2 µg/mL within 4 h after treatment. All four compounds displayed powerful inhibition of the hHDAC activity, with IC
50 values in the range of 0.010–0.212 µM [
165] (
Figure 6).
Figure 6. Chemical structure of other HDAC inhibitors. Diverse chemical structure shows varying range of parasitic HDAC inhibition.
Lately, two recently approved hydroxymate-based HDACis (
36,
51) and four hydroxymate-based HDACis that are already available as anticancer agents under clinical trial were investigated to determine their inhibitory capacity on the proliferation of
T. brucei in the cultured bloodstream. The result of the study revealed that all compounds moderately inhibited the parasite, with EC
50 ranging 0.029 to 11 μM. The most effective compound was
51 (quisinostat) [
166]. Compounds
35 and
36, anticancer drugs, were unable to destroy the cultured parasites at the human-tolerated doses after a single dose administration. Furthermore, compound
36, a sustained-acting HDACi when administered with Human African Trypanosomiasis (HAT) drugs, like pentamidine, suramin, melarsoprol, and nifurtimox, did not display a synergistic effect [
166]. Accordingly, the application of these two HDACis as single agents or in combination for repurposing in HAT treatment is ruled out. Overall, there is lack of associations between the potency against any isoform of hHDAC and the prevention of
T. brucei proliferation. From this idea, the author proposed that HDAC of trypanosome might have a distinctive specificity that could be used for the development of powerful HDACis having selectivity towards the parasite over hHDACs [
166].
2.4. Antitrypanosomal Class-III HDAC (Sirtuin) Inhibitors
Compound
46 has been described as a putative inhibitor of
T. cruzi sirtuin TcSir2rp1 and a growth inhibitor of the parasite at its developmental stages, including trypomastigote and epimastigote. It is evident by a notable inhibition in the number of amastigotes in the
T. cruzi-infected macrophages [
167]. However, besides SIRT inhibition, it also possesses a pleiotropic response; i.e., one gene influences many phenotypic expressions [
167]. Similarly, Moretti et al., characterized two sirtuins, TcSir2rp1 and TcSir2rp3, of
T. cruzi and described that compound
52, an antitumor agent in a different model of cancer, could be a potential SIRTi and inhibit growth and differentiation of
T. cruzi [
79,
111,
168,
169]. Compound
52 diminished the proliferation of epimastigote, with an EC
50 value of 10.6 µM, and prevented its transformation into infective forms. Furthermore, the activity of the recombinant TcSir2rp3 was hampered by compound
52, resulting in an IC
50 value of <1 μM. Hence, it could be proposed that the antiparasitic activity of the compound occurred via TcSir2rp3 inhibition.
Experimental evidence holds up this finding that, with the addition of compound
52, the overexpression of TcSir2rp3 and all phenotypic effects were reduced. Appreciably, compound
52 reduced parasitemia in a
T. cruzi-infected BALB/c mouse model at a lower dose in comparison to a higher in vivo dose that is used for cancer treatment. Nonetheless, compound
52 was not able to prevent the mortality of mice at this dose. Thus, rationally, it can be used to develop a more potent and selective isoform-based SIRTi of the parasite as a potential agent against
T. cruzi infection [
111].
Furthermore, a molecular docking study of pan-SIRTi regarding compounds
42,
46, and SIRTi (thiobarbiturate derivatives) of class-III inhibitors revealed that there was strong correlation between human mitochondrial SIRT5 and TcSir2rp3 with respect to strength and binding mode [
170]. Although the comparison of TcSir2rp3 catalytic pocket with human protein SIRT5 homologous through in silico surface and structural analysis has permitted the recognition of insignificant but notable structural differences at specific inhibitory catalytic domains, that would probably be used for the development of preferable TcSir2rp3 inhibitors with higher specificity towards
T. cruzi [
170].
2.5. Antileishmanial HDAC Inhibitors
The response of
Leishmania parasite towards HDACi indicates the HDACs’ specificity and importance for survivability [
12,
120,
145,
146,
155]. Both antimalarial and antileishmanial properties of hydroxamate-based class-I/II HDACis were discussed by Oyelere et al. [
155]. Furthermore, they studied the effect of 3-hydroxypyridine-2-thiones (3HPTs) on the
L. donovani survivability in amastigote and promastigote forms, which were earlier described as inhibitors of hHDAC6/hHDAC8 [
120]. Compound
3, the pan-HDACi, along with compound
53 (tubastatin A), another hydroxamate-based HDACi, and the reference compound PCI-34051, a selective inhibitor of hHDAC6 and hHDAC8, was taken [
120]. Compound
53, an hHDAC6-selective inhibitor, produced equal antileishmanial activity at both the developmental stages of
L. donovani. This effect was not observed with compound
3 and PCI-34051, a selective hHDAC8 inhibitor. The result also revealed that compounds
54 and
55, 3-HPT-derived HDACi, produced cytotoxicity both at intracellular and extracellular species of
L. donovani, with IC
50 values ranging from 0.1 to 6.5 µg/mL. Thus, the investigator suggested that the antileishmanial efficacy resulted in
L. donovani due to inhibition of HDAC6. This paves a new way for protozoan HDAC6-like activity besides selective isoform inhibitors that might develop a more attentive therapeutic primary plan for Leishmaniasis treatment [
120].
2.6. Antileishmanial Class-III HDAC (Sirtuin) Inhibitors
Still, a smaller number of SIRTis furnished with antileishmanial activity have been described [
12]. Compound
44 demonstrated developmental-stage-specific antiproliferative properties against
L. infantum parasites [
171]. The compound led to apoptotic cell death due to DNA double helix breakage and inhibited the multiplication of axenic amastigotes but did not alter the growth of parasite promastigotes with an IC
50 value of >60 µM [
171]. Overexpression of LmSir2rp1 of parasite results in less susceptibility towards the fragmentation of DNA on the treatment of compound
12 [
171].
Similarly, compound
46 can inhibit the growth of
L. major; even LmSir2rp1 overexpression did not protect the parasite against this compound [
172]. Several in silico and biochemical studies disclosed significant differences between the LmSir2rp1 and catalytic domains of human SIRTs [
102]. In another in vitro investigation, four compounds effectively inhibited the axenic amastigote growth of
L. infantum, but compound
56, a nicotinamide derivative, inhibited LmSir2rp1 [
121]. Later, an investigation revealed that compounds
42 and
46 can also inhibit the recombinant LiSir2rp1, with IC
50 values of 194, and 40 μM, respectively, but similar results were also observed against the human SIRT1 enzyme. Thus, selectivity towards the parasite was lacking [
122]. Furthermore, Tavares et al., described antileishmanial activity and structure–activity relationship study of twelve compounds belonging to bis-naphthalimidopropyl (BNIP) derivatives that varied in the central alkyl chains length, with 2, 3, or 4 nitrogen atoms, connecting two moieties of BNIP [
122]. All derivatives of BNIP could suppress LiSir2rp1, with IC
50 values ranging from 7 to 54.7 μM, and displayed selectivity for hSIRT1 in certain cases. The inhibition and selectivity of LiSir2rp1 seem to rely on linker group length and total charge. Diamine BNIP derivatives having linker 4–7 methylene units were less effective than 8–12 methylene linker units. Further, the additions of the positive amino group to the linker do not affect the selectivity or efficacy.
Compound
57 (BNIP9), having nine methylene units in the linker group, displayed IC
50 value of 5.7 μM against parasitic LiSir2rp1 with 17-fold enhancement in specificity over hSIRT1 [
122]. Furthermore, the derivatives of BNIP could inhibit the intracellular development of
L. infantum amastigotes, with IC
50 values ranging from 1–10 μM. Still, a linear interaction between LiSir2rp1 inhibitory property and antiproliferative effects in the amastigote stage could not be established. Certain derivatives are committed to have favorable antileishmanial efficacy, as shown in
L. infantum-infected BALB/c mouse model [
173].
2.7. Antitoxoplasma HDAC Inhibitors
Several HDACis belonging to class I/II with the cyclic tetrapeptides displayed inhibitory activity on the growth of
Toxoplasma gondii. Compound
5 with IC
50 values ranging from 3 to 15 nM showed its potential but did not possess selectivity over the mammalian cells [
123]. The cyclic tetrapeptide compound
6 obtained from the fermentation broth of
Acremonium species showed rapid growth inhibitory effects against tachyzoites of
T. gondii, with IC
50 value of 10 nM and 13-fold specificity over human foreskin fibroblasts [
132]. These compounds are effective against drug-sensitive and resistant
P. falciparum-infected erythrocytes. Compound
6 caused hyperacetylation of H4 histone of
T. gondii through specific inhibition of TgHDAC3 enzyme. The sensitivity for compounds
5 and
6 was reduced due to single-point mutations within TgHDAC3 (T99A and T99I). Hence, these compounds targeted TgHDAC3, providing genetic proof. Intriguingly, compound
6 displayed turning off the enzyme inhibition due to the insertion of two residues (T99 and A98) within the TgHDAC3 catalytic site. This indicated the presence of a simple safeguard in the HDAC3 family of Apicomplexan, which is not available in any other HDACs of eukaryotes [
132].
In addition, altered gene expression and stage-specific transformation in
T. gondii were completed by TgHDAC3. The treatment with compound
6 converts the replicative tachyzoite to the non-replicative bradyzoite [
174]. A successive study narrates that compound
6 was effective in
T. gondii cysts treated ex vivo and also affected converted cysts and bradyzoites. Particularly, compound
6 diminished the capacity of bradyzoites for conversion to tachyzoites beyond cyst wall injury; thus, isolated free bradyzoites after cell wall lysis were not able to multiply. Ultimately, the cysts formerly treated with compound
6 injected in mice did not produce infection.
Similar studies were conducted for compound
6 cognate (W363 and W399), which is highly active against tachyzoites and afforded better selectivity of 48–62 fold over human foreskin fibroblast cells. Hence, it could be preferable for in vivo studies in the near future [
175]. However, compounds
7,
8, and
9 displayed poor activity against tachyzoites of
T. gondii and showed lower selectivity over HS69 mammalian cells [
135]. Hydroxamate-based HDACis have also been assessed against
T. gondii parasites in the tachyzoite stage [
134]. Among them, compounds
10,
58 (scriptaid), and
3 hampered tachyzoites of
T. gondii, showing IC
50 values of 41, 39, and 83 nM, respectively. However, compound
11 displayed 2–5 times lower potential than others, with IC
50 of 213 nM [
134]. Likewise, compounds
3 and
58 displayed improved selectivity towards the
P. falciparum parasite than compound
10 over mammalian cells [
134]. Surprisingly, compounds
10,
3, and
48 at low concentrations of 1–50 nM reduced the infectivity of
T. gondii tachyzoite through suppression of proliferation and survival [
134]. Very few facts are available on the effectiveness of HDACis belonging to class III against several species of
Toxoplasma. A single report suggested that compound
46 is inactive against tachyzoites of
T. gondii, with an IC
50 value of 50 mM [
134].