1. Cisplatin and Oxaliplatin
Following the FDA approval of cis-diammine dichloroplatinum(II) (cisplatin) in 1978, platinum(II) compounds have been lead chemotherapy drugs. The second-generation drug carboplatin and third-generation drug oxaliplatin are approved worldwide and are used in up to 50% of cancer treatment regimens. The long-term use of Pt(II) drugs in cancer treatment in many patients is limited due to their lack of selectivity to cancer cells causing nephrotoxicity, myelotoxicity and ototoxicity. Between 40 and 80% of patients have experienced permanent hearing loss due to cisplatin. Pt(II) operates as an anticancer agent principally by forming guanine crosslinks with DNA and also monoadducts with guanine
[1], resulting in DNA breaks that ultimately trigger apoptosis. Pt(II) also forms other biomolecular adducts in a non-selective manner in both cancer and noncancer cells, which is a major source of systemic toxicity. Another major limitation of cisplatin is the acquired cellular resistance of cancer cells. Multi-drug resistance proteins are overexpressed to efflux the metal. In this vein, glutathione binds to Pt(II) via its cysteine thiol group and removes the metal through the multidrug resistance protein MRP2. Furthermore, cells activate a number of repair mechanisms, such as the nucleotide excision repair (NER), which repairs DNA and inhibits apoptosis
[1].
Due to an interest in tapping into molecular mechanisms that could trigger ferroptosis in cancer, Guo et al. examined the ferroptosis-inducing capability of traditional chemotherapy drugs in six different cancer cells
[2]. They observed that cisplatin could induce ferroptosis as well as apoptosis in A549 non-small cell lung cancer (NSCLC) and HCT116 human colorectal cancer cells
[2]. It was found that in these cells, Pt(II) significantly decreases the reduced form of glutathione (GSH) via direct coordination to the molecule (
Figure 1). It is known that when cisplatin enters the cell, 60% of it binds to glutathione in the cytoplasm
[3][4]. Typically, as already described, this complexation serves to decrease its cytotoxicity by effluxing the metal, but in these cell lines, the counter effect of ferroptosis appears to outcompete.
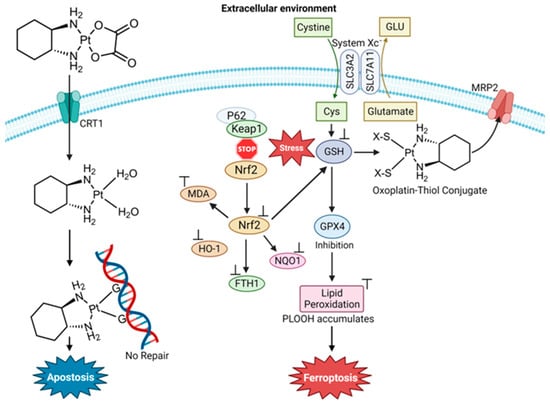
Figure 1. Proposed mechanisms of cisplatin’s induction of apoptosis and ferroptosis (image created using Biorender.com).
Oxaliplatin (
Figure 2) was advanced to the drug market because of its improved safety profile compared to cisplatin and its lack of cross-resistance to cisplatin and carboplatin. The final cytotoxic metabolite of the drug retains the nonhydrolyzable diaminocyclohexane carrier ligand. Similar to cisplatin, oxaliplatin targets DNA by generating guanine cross-links
[5]. Oxaliplatin is a first-line chemo drug for colorectal cancer (CCR). Liu et al. studied the effect of oxaliplatin in HT29 colorectal cancer cells to determine whether it could induce ferroptosis and oxidative stress for insight into new strategies for this highly aggressive cancer
[6]. They found that oxaliplatin triggers ferroptosis by blocking the Nrf2 signaling pathway, resulting in the increase of MDA
[6]. Very importantly, they observed that oxaliplatin enhances the ferroptosis-inducing effect of erastin on CRC cells
[6].
Figure 2. Proposed mechanism of oxaliplatin’s induction of apoptosis and ferroptosis (image created using Biorender.com)
[6].
A recent finding regarding Pt(II) drug blood speciation may also be attributed to the ferroptosis capability of these compounds
[7]. Adult patients receive a standard 90 mg m
−2 dosage of cisplatin. Blood isolated from adults following a round of chemotherapy was found to contain Pt NPs with an average diameter of 6–8 nm and a serum albumin corona of 30 nm in thickness. When the Pt NPs enter into K562 cells, the presence of the mM amount of cytosolic glutathione is capable of dismantling the protein corona, leaving behind the bare Pt NPs
[7]. In this form, the Pt NPs serve as a GSH sponge as the thiol group anchors onto the Pt surface. This GSH anchoring seemingly prevents the GSH-induced efflux of the Pt while also making the GSH biounavailable (
Figure 1).
Currently, efforts are directed at incorporating Pt(II) compounds into different types of anticancer strategies that aim to improve the cancer specificity of the Pt(II). Some strategies include those directed at the development of species that can easily be activated by light and lead to cell death; transition metals being particularly of interest in this area as different types of electronic transitions can be exploited, resulting in longer-term effects and more potent cytotoxicity. Chen et al. developed a degradable NP incorporating a biodegradable Fe(III)-polydopamine (FeP) core with photothermal capability and a hyaluronic acid (HA) crosslinked cisplatin (PtH) shell to achieve a synergistic multifunctional anticancer agent that operates by amplifying intracellular oxidative stress
[8]. PtH@FeP NPs had a ratio of Pt:Fe ranging from 4:3 to 5:3, possibly due to the lack of a reproducible composition. The FeP core facilitated endosomal escape because of the “proton-sponge effect”
[9]. The polydopamines are weakly basic and absorb free protons in the endosome, preventing them from escaping the endosome through diffusion and thus no longer contributing to the internal pH of the endosome. As the absorbed protons accumulate, they cause the membrane potential to increase past the equilibrium level, which results in an influx of chloride and an increase in osmotic pressure. The endosome swells until a critical point at which the lipid bilayer membrane ruptures and releases the endosome contents into the cell. The HA-coated shell enabled the NPs to actively target cancer cells which overexpress the HA receptor. The spherically shaped and negatively charged PtH@FeP NPs were 230 nm in diameter. They enlarge to 602.6 ± 15.2 nm at pH 6.5 attributed to the gelatinous HA shell, which is believed to facilitate long-term drug retention in tumor sites. UV-Vis and IR spectra exhibited the NIR light absorbance of the PtH@FeP NPs. Pt/Fe release from the NPs was investigated under conditions that replicate the extracellular matrix (pH 6.5) and intracellular-reducing environment (10 mM GSH pH 7.4) at 37 °C. Negligible release was observed at pH 6.5. More significant but slow release was observed after treatment with GSH. PtH@FeP showed Pt and Fe cumulative release of 61.43% and 30.21% after 72 h
[8]. The metal release is postulated to be due to the GSH coordination of the Fe(III). It is expected that the PtH@FeP NPs facilitate cytotoxicity through PTT activation at 808 nm laser exposure followed by endosomal release of the NPs. GSH-triggered degradation of the NPs then leads to the released cisplatin attacking DNA and the released labile Fe undergoing Fenton reactivity and ferroptosis onset.
PtH@FeP NP cytotoxicity was examined against 4T1 (murine mammary carcinoma) and HepG2 (hepatocellular carcinoma) cells
[8]. In the case of 4T1, treatment with the NP containing 40 μM Pt and 30 μM Fe and laser application resulted in the almost complete loss of viability after 24 h. In contrast, HepG2 exhibited a nearly 75% viability reduction. Additional characterizations were performed with the 4T1 cell line. Biomarker studies of the NP treatment of the cells showed signature ferroptosis and caspase-dependent apoptosis cell death characteristics. In vivo studies were performed with female Balb/c mice containing 4T1 tumors. The mice were separated into five groups and administered different treatments: PBS, cisplatin, PtH (Pt at 5 mg/kg), PtH@FeP with or without laser (Pt at 3 mg/kg and Fe at 1.8 mg/kg). After 24 h, the tumors were exposed to laser excitation at 808 nm (1 W) for 1 min. Throughout the experiment, the mice’s body weight and tumor volume were closely monitored for 21 days. Cisplatin treatment inhibited tumor growth and even caused a decrease in tumor size with respect to the buffer control, but none of the mice survived after 10 days due to the compound’s toxicity. PtH did not lower the relative tumor volume to the same extent as free cisplatin but it did significantly reduce the compound’s toxicity. There was no significant difference in relative tumor volume between the PtH and PtH@FeP treatment. However, upon irradiation of PtH@FeP, complete suppression of the tumor was observed owing to the synergism of the PTT effect, without any mice’s demise
[8]. Organ histopathology analysis was performed using hematoxylin and eosin (H&E) staining and no organ damage was observed, indicative of the cancer specificity of the system.
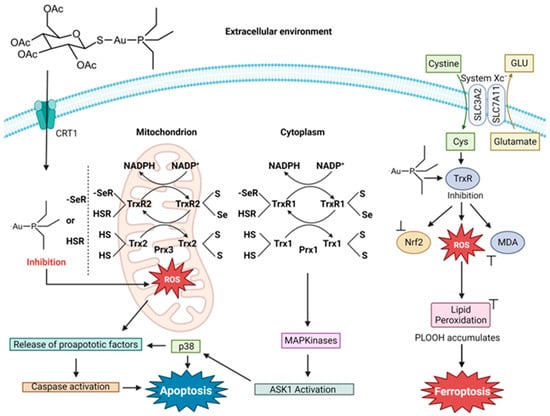
2. Auranofin
Auranofin (AF) was first designed as a therapy for rheumatoid arthritis (RA) and was FDA-approved for this purpose in 1985
[10][11]. Interest in the compound as an anticancer agent stemmed from the decline in cancer progression in certain RA patients being treated with AF. It is a linear Au(I) compound, in which the Au(I) is coordinated to a trialkylphosphine and an S-glycosyl group (2,3,4,6-tetra-
O-acetyl-1-thio-β-d-glucopyranose). The compound is believed to operate as a prodrug like cisplatin
[12] and is capable of inducing cell death in cisplatin-resistant cells
[13]. The sugar group dissociates rapidly and the Au-PEt
3+ complex enters into the cytosol
[14]. Unlike Pt(II) from the Pt(II) drugs, DNA is not the primary target of Au(I). The antiproliferative behavior is believed to be due to the inhibition of thioredoxin (Trx) and thioredoxin reductase (TrxR), which are responsible for regulating redox processes and helping to maintain cellular iron homeostasis
[15]. Au(I) inhibition of the Trx/TrxR system results from binding to their active site thiol and seleno residues
[16][17]. This interaction results in a change in the redox regulation within cells, leading to the excess production of ROS
[16][17]. High ROS levels disturb the activity of cytosolic peroxiredoxin enzymes (Prxs) and activate p38 MAP Kinase (MAPK), which then leads to the initiator caspase triggering the onset of apoptosis. The disturbance of Prxs results in mitochondrial damage and permeability and the release of cytochrome c, turning on its apoptotic function
[17].
The peptide hormone hepcidin, like the Trx/TrxR system, participates in maintaining cellular iron/redox homeostasis
[15]. Hepcidin deficiency can induce iron overload diseases such as hereditary hemochromatosis and anemia. Yang et al. sought to study iron modulation by screening for hepcidin agonists in human hepatic Huh7 cells using a library of over 640 FDA-approved drugs
[18]. They found that AF activates IL-6 signaling, which is responsible for upregulating hepcidin expression. The activation was through the NF-κB pathway. They validated these results in C57BL/6J mice and a mouse model of hemochromatosis (Hfe
−/− mice). Acute treatment of C57BL/6J mice with 5 mg/kg AF decreased the serum Fe by way of activated hepcidin signaling, while chronic treatment of Hfe
−/− mice with 5 mg/kg AF decreased the systemic iron overload in male mice but less so in female mice. Contrary to this beneficial effect, Yang et al. observed that AF is able to induce ferroptosis by way of the inhibition of TrxR activity. Several biomarkers of ferroptosis were detected, including lipid peroxidation. The use of the ferroptosis inhibitor Fer-1 could inhibit the appearance of these biomarkers. In C57BL/6J mice, the role of TrxR inhibition with respect to ferroptosis was revealed through the administration of TRi-1 (25 mg/kg), a specific TrxR inhibitor. Decreased TrxR activity resulted in an accumulation of hepatic lipid peroxidation. By attenuating the ferroptosis capability of AF via the use of Fer-1, AF could exhibit the capacity to decrease the systemic Fe levels. This research reveals how Au(I) activity can be finetuned to turn on and off its ferroptosis induction. For an Fe-based anticancer strategy, turning on Au(I) ferroptosis induction would be very useful (
Figure 3).
Figure 3. Proposed mechanism of auranofin’s induction of apoptosis and ferroptosis (image created using Biorender.com).
Deben et al. sought a deeper understanding of the anticancer mechanism of action of AF and, in particular, its potential application against NSCLC
[19]. This cancer type has a significant elevation of TrxR activity in vitro and in vivo
[20]. Interestingly, high levels of TrxR are associated with chemotherapy resistance, including cisplatin
[21]. Mutant forms of the tumor suppressor protein p53 were found to be sensitizers of AF treatment. Mutant p53 has the potential to alter the cellular redox balance by preventing activation and NF-E2-related factor 2 (NRF2) function. As the main regulator of antioxidant transcription, its inhibition generates an excess of ROS within cancer cells
[22]. Deben et al. worked with NCI-H1299 (p53 null) and its two isogenic derivatives with the accumulation of mutant p53 R175H or R273H. They found that the main mechanism of AF, at both cytostatic (1 μM) and cytotoxic (5 μM) concentrations, is the inhibition of TrxR. The method of cell death induced by AF is dependent on the p53 mutant type. AF sensitized mutant p53 R175H NSCLC cells to caspase-3/7-dependent apoptosis, whereas p53 R273H cells were more vulnerable to ferroptosis
[19]. Regardless of the method of cell death, at a 5 μM concentration, AF depletes GSH. In a study with eight NSCLC and pancreatic ductal adenocarcinoma (PDAC) cell lines with differing p53 status, AF more effectively killed mutant p53 cells with higher p53 protein levels
[19].
3. Photochemotherapeutic Metallodrugs
The practical application of therapeutic treatments for hypoxic and refractory solid tumors has been hindered by their limited efficacy. Photodynamic therapy (PDT) is a treatment option that utilizes a photosensitizing agent and light source to eradicate cancer cells. The utilization of ROS and precise photoirradiation has shown great potential in the struggle against hard-to-treat tumors, resulting in minimal resistance and limited invasiveness. Although ROS cause imprecise damage, the precise photoirradiation allows for a targeted approach. Additionally, PDT offers a diverse range of cell death mechanisms
[23][24].
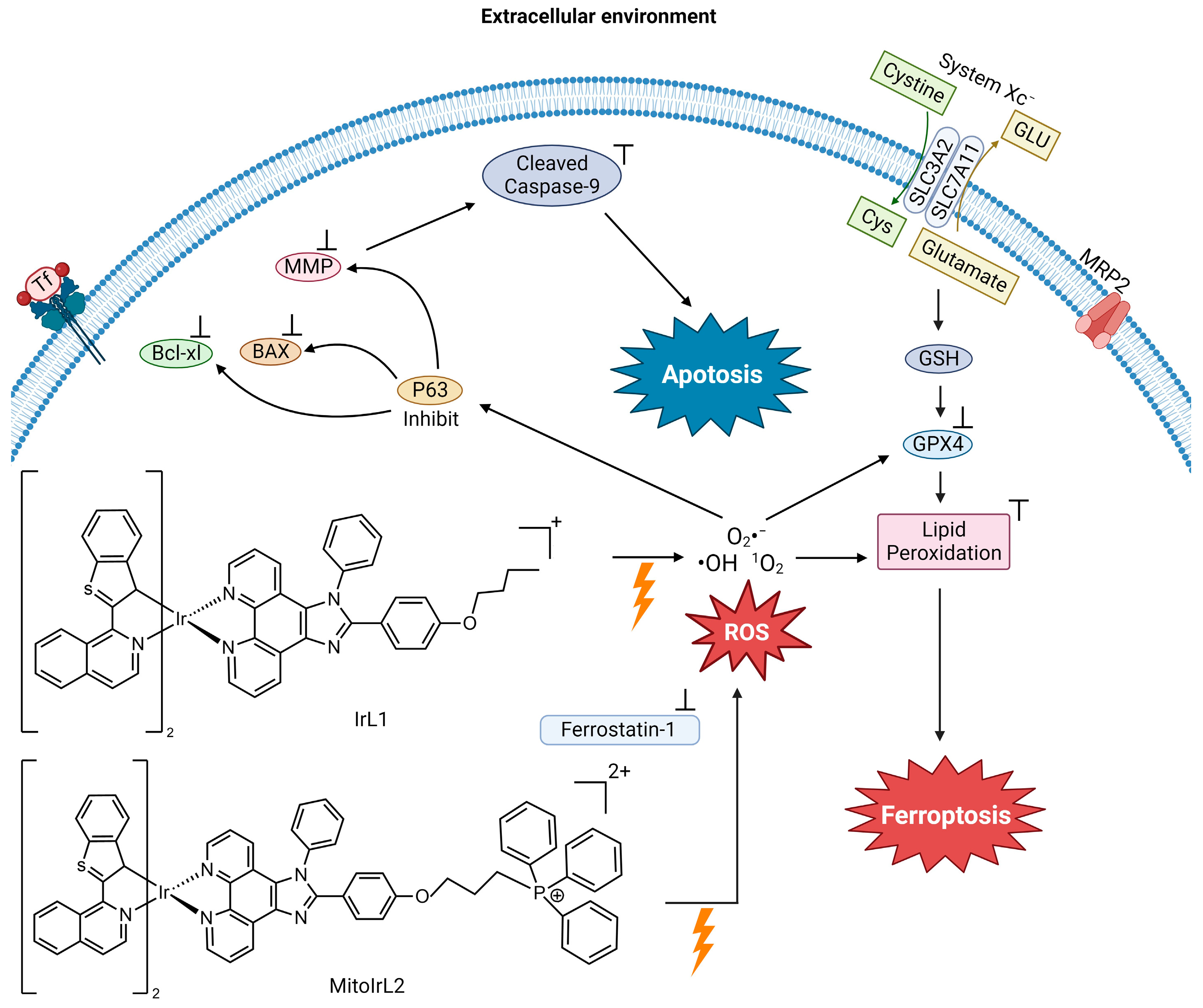
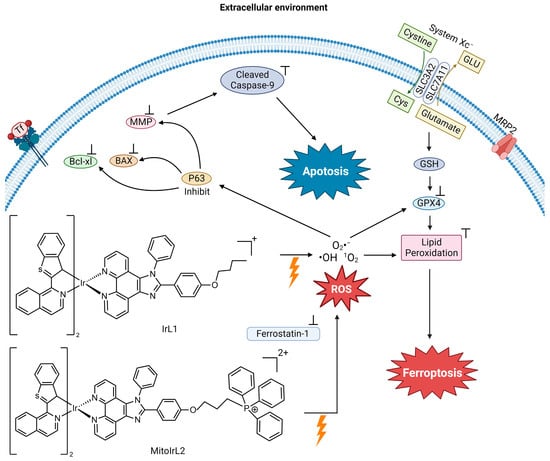
Hao et al. synthesized two Ir(ⅠⅠⅠ) complexes with a coordination number 6, containing two 1-benzo[b]thien-2-yl-isoquinoline (btiq) ligands and an imidazophenanthroline (ipt) auxiliary ligand; IrL1 and MitoIrL2 (
Figure 4) to examine their use as PDT agents for hypoxic cells
[25]. Both complexes exhibit intensive absorption bands at ~488 nm due to a metal-to-ligand charge transfer (MLCT). The complexes display a near-IR emission at ~685 nm, with a shoulder at 740 nm. The emission intensity decreases in the presence of ambient O
2, indicative of excited forms of the compounds possibly being able to undergo intersystem crossing (ISC) to react with O
2 and produce ROS. Given these physicochemical properties, the complexes were studied as photodynamic therapy (PDT) agents. They analyzed the ROS species formed in Michigan Cancer Foundation-7 (MCF-7) breast cancer cells in two different biological environments. These are normoxia (an environment with normal levels of oxygen) and hypoxia (an environment with a reduction in oxygen) because they expected that these compounds would be able to overcome the resistance of cells in a hypoxic environment. The IrL1 and MitoIrL2 complexes at 5 μM were photoirradiated at 450 nm (30 Jcm
−2). In a normoxia environment, they produce singlet oxygen (
1O
2), but not in the hypoxic environment. An increase in the ROS species O
2− and ·OH were observed under both conditions, but MitoIrL2 produced a greater amount
[25]. At a concentration of 1 μM of both complexes, the GPX4 levels decreased and lipid peroxide accumulated
[25].
Figure 4. Proposed mechanism of IrL1 and MitoIrL2’s induction of apoptosis and ferroptosis (image created using Biorender.com)
[25].
The mechanism of cell death was determined by incorporating the inhibitors of apoptosis (z-VADfmk), necrosis (necrostatin-1, Nec-1), autophagy (3-methyladenine, 3-MA) and ferroptosis (Fer-1) under hypoxic conditions in a cell viability assay
[25]. Fer-1 addition produced an increase in cell viability in treatments with both complexes, indicating that both induce ferroptosis. The z-VADfmk inhibitor increased the cell viability in treatment with MitoIrL2, suggesting that the compound also induces apoptosis. For this compound, apoptosis is the dominant cell death mechanism. According to flow cytometry, IrL1 caused apoptotic cell death in 9.5% of the total dead cells, whereas MitoIrL2 caused 43.1%. Western blot assays were carried out to assess apoptosis biomarkers. MitoIrL2 increased the p53 and Bax levels, while the Bcl-xl levels decreased, leading to a rise in caspase-9
[25]. The compound was observed to inhibit ATP production and cause mitochondrial structural changes. This research elucidates how the finetuning of the physicochemical properties of a PDT agent can increase the cytotoxic potency of a metal compound and facilitate dual cell death mechanisms.
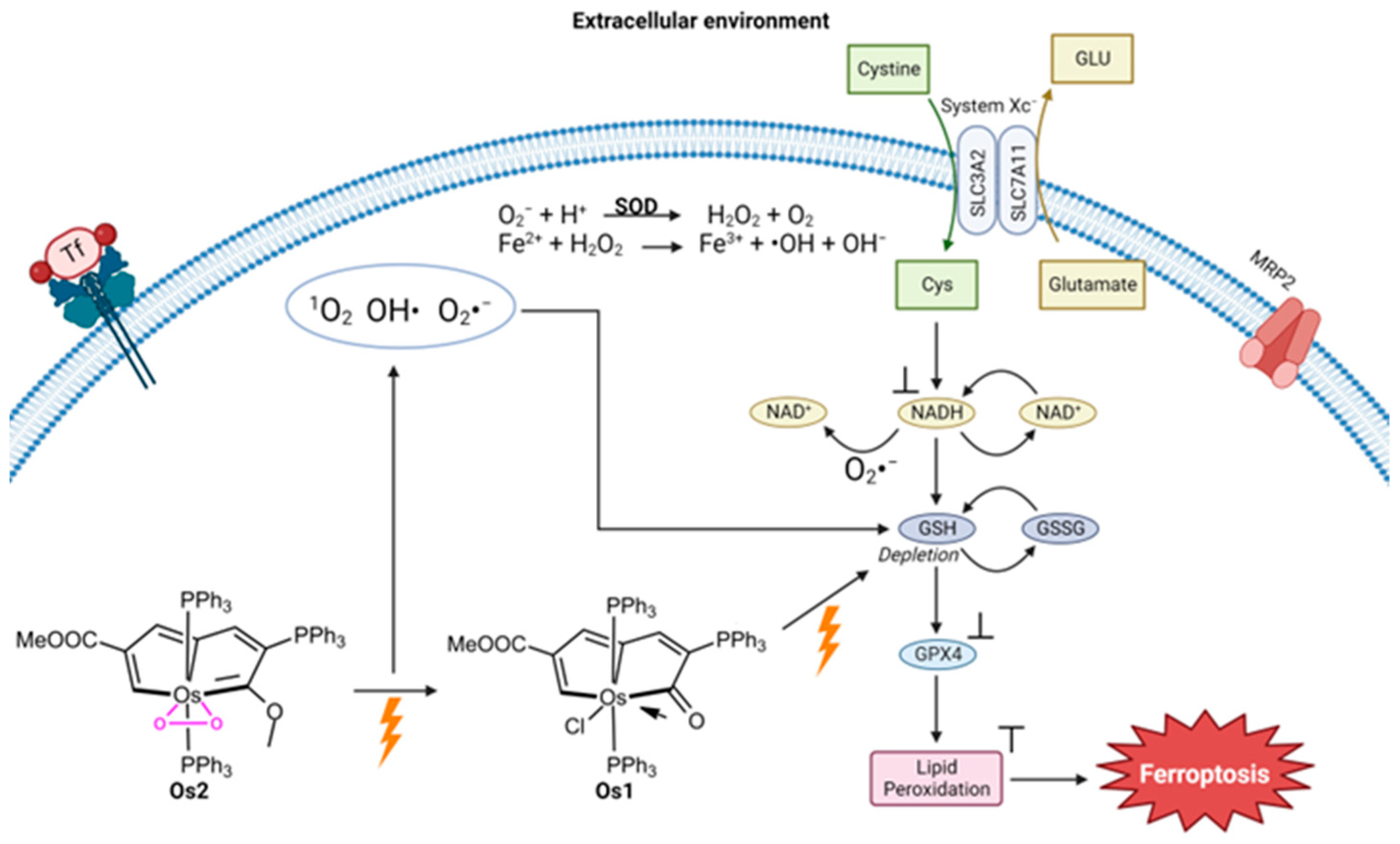
Another metal that has been studied for its ability to trigger ferroptosis via photodynamic therapy (PDT) is osmium. Zhang et al. investigated a way to treat hypoxic cancer cells using a pentagonal bipyramidal complex of Os (Os2) containing pentalyne and peroxo ligands in the equatorial plane and two triphenylphosphine ligands in the axial position
[26]. Using photoirradiation under light illumination (465 nm, 13 mW/cm
2), Os2 releases its peroxo ligand and converts it into Os1 (
Figure 5) and the superoxide (O
2−) ROS. An ROS detection experiment was performed with HeLa cervical cancer cells treated with Os2 under hypoxic conditions and light irradiation. The generation of O
2− was observed as well as OH and
1O
2. Under normal oxygen conditions, exposure to Os2 and Os1 results in considerable cell death following irradiation, with the viability of HeLa cells dropping below 20%. Similar results are observed in hypoxia, although the degree of cell death is reduced at lower concentrations, both before and after irradiation, compared to normoxia. After administering Os2 treatment, the GSH percentage in cells was examined and the results showed that the GSH levels in the irradiated group were significantly lower than those in the non-irradiated group. The relative expression levels of GPX4 were lower post-irradiation, and in fact, were at the similar level as HeLa cell treatment with the ferroptosis inducer RSL3. The photocatalytic oxidation of NADH into NAD+ was observed by Os2 under light irradiation, which is characteristic of ferroptosis induction.
Figure 5. Proposed mechanism of Os2’s induction of ferroptosis (image created using Biorender.com)
[26].
An in vivo study was conducted by treating Balb/c mice implanted with HeLa tumors
[26]. Following 7 days after cell injection, mice with a desired tumor volume (~100 mm
3) were selected for further study. The mice were separated into four groups: control dark group with only PBS injection, control light irradiation group, Os2-dark group, and Os2-light irradiation group. The irradiation (465 nm with an intensity of 13 mW/cm
2 for a duration of 60 min) was performed on day zero and the tumor size was recorded every two days. It was observed that the Os2-light irradiation group had the lowest growth in tumor size, roughly 2.33 times less growth than the control dark group. Biosafety analysis of the Os compound was performed by injecting healthy mice with three times the therapeutic dose and performing H&E staining of thin slices of various organs. No tissue damage was observed. Toxicity was also examined in zebrafish and no blood vessel damage was detected. These results attest to the cancer specificity and low toxicity of the Os2 system.
4. Conclusions
Small molecule non-iron metal complexes demonstrate both the importance of the metal center and the physicochemical properties of intact metal–ligand adducts. The classical Pt(II) and Au(I) drugs reveal how soft Lewis acid metal ions can strongly bind to thiol and seleno proteins and ablate redox regulation, leading to ferroptosis. In the case of Au(I), the particularities of the tumor biochemical environment, such as the presence of particular mutant P53 proteins, dictate whether Au(I) is able to turn on ferroptosis
[19]. Some metal complexes that are light sensitive demonstrate photosensitizing and photothermal capability to facilitate tumor-localized applications that can be coupled with ferroptosis-inducing agents. Photosensitizing metal complexes demonstrate the utility of PDT to enable the generation of ROS and
1O
2 that can synergize with intracellular Fe to produce LPO. PTT agents can be synergized with redox active metals to facilitate more than one type of death. Collectively, these studies reveal that it is highly unlikely that any drug strategy can purely induce ferroptosis alone, which is not a bad thing. The synergism of approaches to trigger multiple cell death pathways may prove vital for the drug development of far more potent and less cancer-resistant chemotherapy.
 +1 credit
+1 credit