 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | thomas vaccari | -- | 2046 | 2024-01-22 09:35:55 | | | |
| 2 | Sirius Huang | Meta information modification | 2046 | 2024-01-23 02:14:17 | | |
Video Upload Options
Protein homeostasis is essential for neuron longevity, requiring a balanced regulation between protein synthesis and degradation. The clearance of misfolded and aggregated proteins, mediated by autophagy and the ubiquitin–proteasome systems, maintains protein homeostasis in neurons, which are post-mitotic and thus cannot use cell division to diminish the burden of misfolded proteins. When protein clearance pathways are overwhelmed or otherwise disrupted, the accumulation of misfolded or aggregated proteins can lead to the activation of ER stress and the formation of stress granules, which predominantly attempt to restore the homeostasis by suppressing global protein translation. Alterations in these processes have been widely reported among studies investigating the toxic function of dipeptide repeats (DPRs) produced by G4C2 expansion in the C9orf72 gene of patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).
1. Introduction
2. The Impact of C9-Associated Toxic Repeats on Protein Degradation Pathways
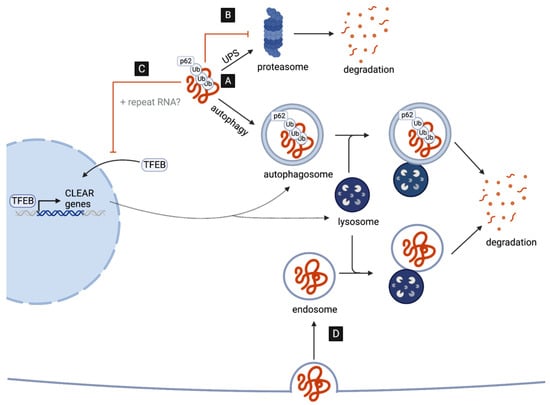
2.1. Poly-GA Inhibits Protein Degradation via the UPS
2.2. C9-Associated Toxic Repeats Disrupt Autophagosome and Lysosome Biogenesis
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in als and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42.
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268.
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256.
- Byrne, S.; Heverin, M.; Elamin, M.; Walsh, C.; Hardiman, O. Intermediate repeat expansion length in C9orf72 may be pathological in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 148–150.
- Chen, Y.; Lin, Z.; Chen, X.; Cao, B.; Wei, Q.; Ou, R.; Zhao, B.; Song, W.; Wu, Y.; Shang, H.F. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 38, e15–e217.e22.
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115.
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447.
- Fratta, P.; Poulter, M.; Lashley, T.; Rohrer, J.D.; Polke, J.M.; Beck, J.; Ryan, N.; Hensman, D.; Mizielinska, S.; Waite, A.J.; et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013, 126, 401–409.
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.C.; Vieira De Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438.
- Zhang, Y.J.; Jansen-West, K.; Xu, Y.F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524.
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.I.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645.
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grässer, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014, 128, 485–503.
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154.
- Todd, T.W.; McEachin, Z.T.; Chew, J.; Burch, A.R.; Jansen-West, K.; Tong, J.; Yue, M.; Song, Y.; Castanedes-Casey, M.; Kurti, A.; et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration In Vivo. Cell Rep. 2020, 31, 107616.
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443.
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125.
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194.
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133.
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013, 126, 845–857.
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA Foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186.
- Cooper-Knock, J.; Higginbottom, A.; Stopford, M.J.; Highley, J.R.; Ince, P.G.; Wharton, S.B.; Pickering-Brown, S.; Kirby, J.; Hautbergue, G.M.; Shaw, P.J. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015, 130, 63–75.
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977.
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844.
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; Van Der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893.
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338.
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; DeJesus-Hernandez, M.; Van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646.
- Mann, D.M.A.; Rollinson, S.; Robinson, A.; Bennion Callister, J.; Thompson, J.C.; Snowden, J.S.; Gendron, T.; Petrucelli, L.; Masuda-Suzukake, M.; Hasegawa, M.; et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2014, 2, 68.
- Schludi, M.H.; May, S.; Grässer, F.A.; Rentzsch, K.; Kremmer, E.; Küpper, C.; Klopstock, T.; Ceballos-Baumann, A.; Danek, A.; Diehl-Schmid, J.; et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015, 130, 537–555.
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 2018, 23, 1–12.
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696.e12–705.e12.
- Khosravi, B.; LaClair, K.D.; Riemenschneider, H.; Zhou, Q.; Frottin, F.; Mareljic, N.; Czuppa, M.; Farny, D.; Hartmann, H.; Michaelsen, M.; et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020, 39, e102811.
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677.
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 2017, 4, ENEURO.0249-16.2017.
- Zhang, Y.; Nelson, S.C.K.; Viera Ortiz, A.P.; Lee, E.B.; Fairman, R. C9orf72 proline-arginine dipeptide repeats disrupt the proteasome and perturb proteolytic activities. J. Neuropathol. Exp. Neurol. 2023, 82, 901–910.
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278.
- Van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE 2011, 6, e22850.
- MacKenzie, I.R.; Arzberger, T.; Kremmer, E.; Troost, D.; Lorenzl, S.; Mori, K.; Weng, S.M.; Haass, C.; Kretzschmar, H.A.; Edbauer, D.; et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol. 2013, 126, 859–879.
- Gomez-Deza, J.; Lee, Y.B.; Troakes, C.; Nolan, M.; Al-Sarraj, S.; Gallo, J.M.; Shaw, C.E. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol. Commun. 2015, 3, 38.
- Baborie, A.; Griffiths, T.D.; Jaros, E.; Perry, R.; McKeith, I.G.; Burn, D.J.; Masuda-Suzukake, M.; Hasegawa, M.; Rollinson, S.; Pickering-Brown, S.; et al. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol. Appl. Neurobiol. 2015, 41, 601–612.
- Vatsavayai, S.C.; Yoon, S.J.; Gardner, R.C.; Gendron, T.F.; Vargas, J.N.S.; Trujillo, A.; Pribadi, M.; Phillips, J.J.; Gaus, S.E.; Hixson, J.D.; et al. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 2016, 139, 3202–3216.
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458.
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500.
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477.
- Cunningham, K.M.; Maulding, K.; Ruan, K.; Senturk, M.; Grima, J.C.; Sung, H.; Zuo, Z.; Song, H.; Gao, J.; Dubey, S.; et al. Tfeb/mitf links impaired nuclear import to autophagolysosomal dysfunction in c9-als. eLife 2020, 9, e59419.
- Sung, H.; Lloyd, T.E. Disrupted endoplasmic reticulum-mediated autophagosomal biogenesis in a Drosophila model of C9-ALS-FTD. Autophagy 2023, 20, 94–113.
- Wang, S.M.; Wu, H.E.; Yasui, Y.; Geva, M.; Hayden, M.; Maurice, T.; Cozzolino, M.; Su, T.P. Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy 2023, 19, 126–151.
- Zhang, K.; Grima, J.C.; Rothstein, J.D.; Lloyd, T.E. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus 2016, 7, 132–137.
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61.
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140.e11.
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117.
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774.
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433.
- Bouché, V.; Espinosa, A.P.; Leone, L.; Sardiello, M.; Ballabio, A.; Botas, J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy 2016, 12, 484–498.
- Tognon, E.; Kobia, F.; Busi, I.; Fumagalli, A.; De Masi, F.; Vaccari, T. Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy 2016, 12, 499–514.
- Bozič, J.; Motaln, H.; Janez, A.P.; Markič, L.; Tripathi, P.; Yamoah, A.; Aronica, E.; Lee, Y.B.; Heilig, R.; Fischer, R.; et al. Interactome screening of C9orf72 dipeptide repeats reveals VCP sequestration and functional impairment by polyGA. Brain 2022, 145, 684–699.
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676.
- Yang, M.; Chen, L.; Swaminathan, K.; Herrlinger, S.; Lai, F.; Shiekhattar, R.; Chen, J.F. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016, 2, e1601167.
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51.
- Corbier, C.; Sellier, C. C9ORF72 is a GDP/GTP exchange factor for Rab8 and Rab39 and regulates autophagy. Small GTPases 2017, 8, 181–186.
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408.
- Wang, M.; Wang, H.; Tao, Z.; Xia, Q.; Hao, Z.; Prehn, J.H.M.; Zhen, X.; Wang, G.; Ying, Z. C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell 2020, 19, e13126.
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574.
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 2018, 2018, e5815.
- Marchi, P.M.; Marrone, L.; Brasseur, L.; Coens, A.; Webster, C.P.; Bousset, L.; Destro, M.; Smith, E.F.; Walther, C.G.; Alfred, V.; et al. C9ORF72-derived poly-GA DPRs undergo endocytic uptake in iAstrocytes and spread to motor neurons. Life Sci. Alliance 2022, 5, e202101276.
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652.
- Chang, Y.J.; Jeng, U.S.; Chiang, Y.L.; Hwang, I.S.; Chen, Y.R. The glycine-alanine dipeptide repeat from C9 or f72 hexanucleotide expansions forms toxic amyloids possessing cell-to-cell transmission properties. J. Biol. Chem. 2016, 291, 4903–4911.
- Zhou, Q.; Lehmer, C.; Michaelsen, M.; Mori, K.; Alterauge, D.; Baumjohann, D.; Schludi, M.H.; Greiling, J.; Farny, D.; Flatley, A.; et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol. Med. 2017, 9, 687–702.

